Heterogeneous phenotype of autoinflammatory disease in a patient with mutations in NOD2 and MEFV genes
Abstract
Background: Autoinflammatory diseases are a genetically heterogeneous group of conditions characterized by excessive activation of the innate immune system. They frequently present with overlapping features, particularly in cases of digenic or polygenic inheritance. The most common cause of autoinflammation arises from causative variants in the MEFV gene, responsible for familial Mediterranean fever. Clinical features include recurrent episodes of fever with serositis and amyloidosis. Individuals with variants in MEFV that present atypically with heterogeneous autoinflammatory features have also been described. Notably, gene modifiers of MEFV, such as NOD2 encoding an intracellular bacterial sensor, can result in more severe disease. NOD2 underlies a number of autoinflammatory and immunodeficiency conditions, including Blau syndrome. To date, Blau syndrome has not been described in the context of MEFV.
Aim: To expand the presentation and phenotype of autoinflammatory disease associated with defects in the NOD2 and MEFV genes.
Methods: A retrospective review of the patient’s chart was performed, including family history, medical history, immune laboratory evaluation, and genetics.
Results: We describe here a 68-year-old male with a remarkable medical history since childhood of skin rash, erythroderma, recurrent infections, autoinflammation, arthritis, uveitis, and malignancy. A significant family history of cancer and autoinflammation was noted. Genetic work-up involving a 17-gene autoinflammatory panel revealed 3 heterozygous variants of uncertain significance, 2 of which were present in the MEFV gene and one in the NOD2 gene. His features were consistent with an overlapping phenotype of Blau syndrome and atypical FMF.
Conclusion: Heterozygous variants in NOD2 and MEFV can result in a spectrum of autoinflammatory disorders with a heterogeneous phenotype. The NOD2 variant identified in our patient has not previously been associated with Blau syndrome.
Statement of Novelty: We describe a patient harbouring heterozygous mutations in the MEFV and NOD2 genes marked by recurrent childhood infections.
Introduction
Autoinflammatory diseases are a genetically heterogeneous group of conditions characterized by excessive activation of the innate immune system, and are distinguished from autoimmunity by the absence of autoantibodies and non-dependence on antigens (McGonagle and McDermott 2006; Martorana et al. 2017; Nigrovic et al. 2020). Aberrations in innate immune function, such as those involved in the detection of pathogens and (or) downstream signalling pathways that relay these responses, form the basis of the 5 major categories of autoinflammatory diseases. These include (i) inflammasomopathies (defects in multi-protein inflammasomes responsible for caspase-1-dependent IL-1β production), (ii) interferonopathies (defects in intracellular DNA sensors or counteracting nucleases leading to constitutive production of type I interferons), (iii) unfolded protein/cellular stress response syndromes (defects in handling of misfolded proteins resulting in activation of immune cascades, such as inflammasomes and interferon pathways), (iv) relopathies (defects in regulators of NF-κB-activity leading to hyperactivation), and (v) uncategorized (Rood and Behrens 2022). The clinical features frequently overlap and may include fever, rash, serositis, meningitis, gastrointestinal manifestations, and arthritis. A diagnosis is made based on correlation of symptoms against those of known autoinflammatory diseases (Tunca and Ozdogan 2005) and may be supported by genetic analysis (Simon et al. 2006).
With the advancement of genetic sequencing techniques, this field has grown exponentially with now recognized monogenic and polygenic origins (Pathak et al. 2017). Manifestations of digenic inheritance arising from the MEFV gene, causing the most common form of autoinflammatory disease – familial Mediterranean fever (FMF), alongside the NOD2 gene, have been previously noted (Yao et al. 2019).
The MEFV gene is located on chromosome 16 and encodes pyrin, a 781-amino acid protein that serves as a cytosolic sensor of bacterial-induced Rho GTPase inactivation. Pyrin is expressed in the cytoplasm of cells from myeloid lineages, synovial fibroblasts, and dendritic cells. Engagement and assembly of the pyrin inflammasome activates caspase-1, resulting in proinflammatory cytokine IL-1β and IL-18 production, and pyroptosis (inflammatory cell death) (Nigrovic et al. 2020). Autosomal recessive and autosomal dominant mutations in MEFV, causing excess activation of the pyrin inflammasome, can result in the clinical phenotype of FMF (OMIM # 134610, #249100) (Booty et al. 2009; Chae et al. 2011; Nigrovic et al. 2020; Tunca and Ozdogan 2005). While FMF can affect any ethnic group, the prevalence is estimated to be as high as 1:200 in certain Mediterranean populations.
Presentation of FMF usually occurs during childhood with major criteria of recurrent episodes of fever with serositis, amyloidosis and (or) favorable response to colchicine, and minor criteria encompassing recurrent fever, erysipelas-like erythema, or a first degree relative with FMF. Other features include abdominal and (or) chest pain, monoarticular arthritis, neutrophilia, elevated inflammatory markers, and amyloidosis commonly involving the kidneys.
A role for NOD2 as a gene modifier for FMF, causing more severe disease, has been suggested (Berkun et al. 2012). The NOD2 gene is located on chromosome 16 and encodes NOD2 (nucleotide-binding oligomerization domain-containing protein 2), a member of the intracellular NOD-like receptor (NLR) family of pattern recognition receptors (PRR). This innate sensor detects bacterial motifs (peptidoglycan) and promotes bacterial clearance through activation of the NF-kB pathway and induction of autophagy (Caruso et al. 2014). NOD2 is expressed primarily in monocytes but is also present in macrophages, dendritic cells, lymphocytes, epithelial and endothelial cells, as well as intestinal Paneth cells.
Defects in NOD2 are categorized as relopathies, due to dysregulated activation of the NF-kB pathway (Negroni et al. 2018; Steiner et al. 2018; Mukherjee et al. 2019). To date, pathogenic mutations in NOD2 have been associated with Blau Syndrome (OMIM 186580)(Miceli-Richard et al. 2001)/Early Onset Sarcoidosis, Crohn’s Disease (OMIM 266600)(Ogura et al. 2001; Hugot et al. 2001; King et al. 2006), and Yao Syndrome (OMIM 617321) (Yao and Shen, 2017). Conditions in this group involve autoinflammation with or without primary immunodeficiency traits (Pathak et al. 2017; Kacar et al. 2019).
Here, we report the complex medical and family history of an adult male who presented at an early age with autoinflammation and immunodeficiency. Targeted panel sequencing identified 3 missense mutations, 2 in MEFV and 1 in NOD2.
Case presentation
Our patient is currently a 68-year-old male of European descent who was referred to our clinic at age 64 after a remarkable and complex medical history since childhood.
Family history
The patient’s family history is notable for malignancy with prostate, bowel, and laryngeal cancer in his father, bowel cancer in his paternal grandmother, and breast cancer in his paternal aunt. He had a sister who died before the age of 1 year from an unknown cause, and his brother has a history of viral cardiomyopathy. His mother had coronary artery disease, Sick Sinus Syndrome and recurrent episodes of pulmonary embolism and deep vein thrombosis. His father, in addition to malignancy, also had Sick Sinus Syndrome. His daughter has hypothyroidism and his son has Crohn’s disease.
Infections
Our patient presented with repeated tonsillitis from 4 years of age and recurrent sinus infections from 10 years of age, which required surgery and repeated admission for intravenous antibiotics even through to adulthood. At age 7 years, he had right ulnar osteomyelitis requiring surgical management and plate insertion during early adulthood. Other infections included recurrent viral infections, Herpes Zoster, and recurrent warts since the age of 40 years. At 46 years of age, he was admitted for intravenous antibiotic therapy due to prostatitis and septicemia. He also had multiple episodes of cellulitis since childhood, mainly on his arms and hands with increased frequency during the last few of years, including repeated cyst sebaceous infection. Biopsy confirmed dermal scarring reaction with abscess formation including prominent foreign body type granulomatous reaction. Another skin lesion of interest was a perianal condyloma found on pathology.
Gastroenterology
He suffered from recurrent abdominal pain, diarrhea, and bloating since early adulthood. After several evaluations during his 50´s, he was found to have bowel polyps, adenomas, and lymphoid hyperplasia. A liver biopsy showed mild steatosis and minimal fibrosis. At age 54 years, he was diagnosed with sclerosing mesenteritis and is currently treated with colchicine, tamoxifen, steroids, and methotrexate. In his late 60’s, he developed pancreatitis due to a pseudocyst, complicated with acute renal failure. No hepatosplenomegaly is noted.
Autoinflammation and autoimmunity
At the age of 44 years, autoimmune thyroiditis and hypothyroidism were identified. He also experienced triphasic Raynaud’s phenomenon, which worsened during winter. By his late adulthood he was diagnosed with iritis, a form of uveitis, which concurred with a Herpes Zoster infection. By 63 years of age, he had 7 hospitalizations including 1 for possible viral encephalitis, mitochondrial disease, or cryoglobulinemia. He has cervical osteoarthritis affecting his vertebral column which is managed by steroids.
Cardiovascular
During his late 50’s, our patient was diagnosed with mild aortic root dilatation and left ventricular hypertrophy with hypertension, and in his early 60’s with premature ventricular contractions, bigeminy, trigeminy, palpitations, and bundle branch block. He developed severe mitral valve regurgitation at 61 years old. During that time he presented with an unprovoked deep vein thrombosis, and remains on long-term treatment with warfarin. On recent evaluation, a grade 3/6 pan-systolic murmur and trigeminal arrhythmia were noted.
Malignancy
Following 10 years of severe prostatitis, he was diagnosed at age 56 years with prostate cancer and underwent radical prostatectomy. He has since had 2 normal bone marrow aspirates with no signs of lymphoproliferative disorders.
Neurologic
At 52 years of age, concerns about transient ischemic attack were raised after sudden unexplained and non-specific weakness of his right upper extremity. MRI showed small chronic left cerebellar infarct and chronic small vessel ischemic background changes. Upon psychiatric evaluation, cortical problems, decreased performance, and concentration were noted.
Respiratory
The patient has suffered from severe obstructive sleep apnea since age 60 years. A CT scan of the chest revealed 2 lung nodules with some atypical fibrosis.
Physical evaluation
The patient has facial plethora and flush, bilateral supraclavicular lymphadenopathy, and his ear, nose, and throat demonstrate edematous nasal turbinate. The skin on his upper back has some round scaly, shiny lesions of 2-3mms in diameter, which the patient describes as initially appearing as a red bump, then turning white at the centre and becoming scaly. He has multiple telangiectasias on his arms and back as well as a baseline erythroderma.
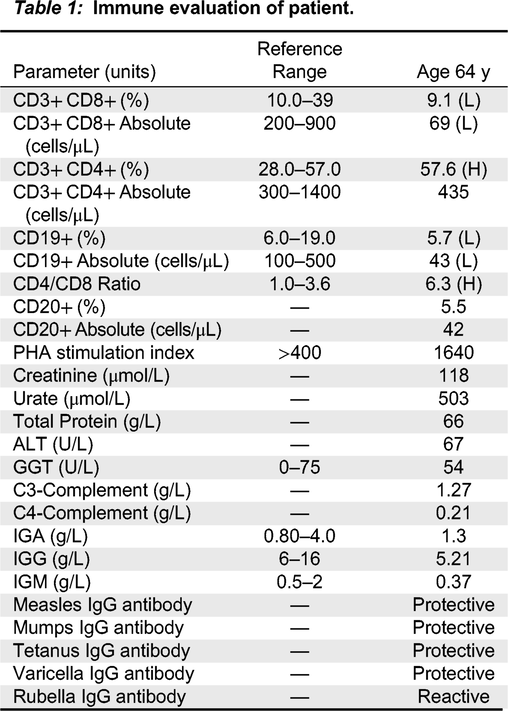
Immune evaluation
Immune investigations performed in the context of prolonged use of steroids showed slightly low immunoglobulins G and M, with normal Immunoglobulin A (IgG: 5.21 g/L, IgA 1.4 g/L, IgM: 0.37 g/L) while specific antibody titers against mumps, measles, rubella, tetanus, and varicella were protective (Table 1). Lymphocyte immunophenotyping demonstrated low B cell numbers of 43 cells/μL (normal: 100–500 cells/μL), low CD8+ T cells (CD3+CD8+: 69 cells/μL, normal: 200–900 cells/μL; CD3+CD4+: 435 cells/μL, normal: 300–1400 cells/μL) with a significant predominance of CD4+ over CD8+ T cells, at a ratio of 6.3 (normal: 1–3.6). Lymphocyte proliferation to the mitogen phytohemagglutinin (PHA) was normal, with a stimulation index of 1649. Autoantibodies were weakly positive for anti-Ro 52 and anti-SCL 70/topoisomerase. He receives IVIG based on borderline low IgG and IgM levels.
Table 1:
Genetic investigations
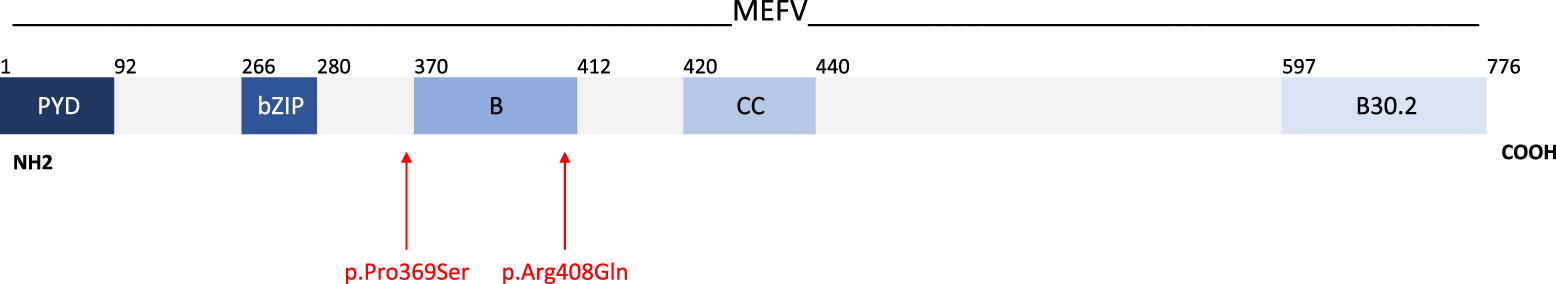
A 17-gene autoinflammatory disease panel detected 3 variants of uncertain significance. A heterozygous variant in the NOD2 gene (NM_022162.1)(Figure 1), resulting from the substitution of an arginine residue with tryptophan at position 702, NOD2:c.2104C>T (p.Arg702Trp), was reported. While there is no consensus regarding the pathogenicity of this variant (Sift, Polyphen, Mutations Taster), it has previously been reported in patients with features of Yao Syndrome (Yao et al. 2011; Yao and Shen 2017; Chapman et al. 2009) as well as Crohn’s disease (Yazdanyar et al. 2009; Chapman et al. 2009; Christodoulou et al. 2013). Additionally, 2 heterozygous variants in MEFV gene (NM_000243.2)(Figure 2) were noted, 1 resulting in the substitution of proline with serine in the position 369, MEFV:c.1105C>T (p.Pro369Ser), with no consensus regarding its pathogenicity (Aksentijevich et al. 1999; Hannan et al. 2012; Migita et al. 2014), and the other resulting in the substitution of an arginine with a glutamine residue at position 408, MEFV:c.1223G>A (p.Arg408Gln), predicted to be benign (Cazeneuve et al. 1999; Ryan et al. 2010; Hannan et al. 2012; Migita et al. 2014). Both MEFV variants have a high frequency in control populations and been documented as low penetrance mutations in cases of FMF. The combination of these 2 MEFV gene variants have been reported in multiple individuals with FMF with a highly variable phenotype (Cazeneuve et al. 1999; Ryan et al. 2010).
Figure 1:

Figure 2:
Discussion
Here, we describe a patient who presented with 3 heterozygous variants of uncertain significance, 2 in the MEFV gene (associated with autoinflammatory disease) and 1 in the NOD2 gene (associated with autoinflammatory disease and immunodeficiency).
The diagnosis of FMF in adults is based on the fulfillment of either 2 or more major criteria of recurrent episodes of fever with serositis, amyloidosis, favorable response to colchicine, or 1 major and 2 minor criteria of recurrent fever, erysipelas-like erythema, or a first degree relative with FMF. Our patient’s clinical phenotype did not fit the classical phenotype of FMF, given the absence of symptoms listed above. Yet, a case series by Ryan and colleagues (Ryan et al. 2010) previously reported the heterogenous and atypical FMF phenotype of a cohort of patients harbouring heterozygous MEFV mutations, including the identical 2 mutations that were present in our patient, c.1105C>T (p.Pro369Ser) and c.1223G>A (p.Arg408Gln). Both are located within exon 3 of MEFV encoding the B-box zinc finger domain (Figure 2), a region that negatively regulates activation of the pyrin inflammasome. While 17 of 22 patients did not meet the diagnostic criteria for FMF, a clustering of symptoms was reported, including 1 whose symptoms included panuveitis, inflammatory bowel disease, lethargy, and intermittent fever. Lymphadenopathy and lack of response to colchicine, the mainstay treatment for FMF, were noted in a number of patients. These symptoms overlap with our patient’s presentation (with the exception of fever) and may partially explain his complex phenotype.
The coexistence of MEFV and NOD2 genetic variants, both located in chromosome 16, is relatively common and is marked by overlapping clinical features, variable penetrance, leading to phenotypic heterogeneity and therapeutic variability (Yao et al. 2019). Case reports and cohorts of such combinations have demonstrated a high association with Crohn’s disease among patients with FMF (Berkun et al. 2012). The NOD2 mutation present in our patient, c.2104C>T (p.Arg702Trp), is considered a risk factor for Crohn’s disease, and has also been described in patients with Yao Syndrome, an autoinflammatory disease characterized by periodic fever, dermatitis, arthritis, swelling of the distal extremities, gastrointestinal and sicca-like symptoms, and serositis (Yao and Shen, 2017; Yao et al. 2019). Notably, the phenotype of our patient was not consistent with either Crohn’s disease or Yao syndrome, particularly given the lack of fever — a major criteria for Yao Syndrome. Moreover, although there is often clinical overlap between Yao Syndrome and FMF, with some case reports suggesting that patients may present features of both autoinflammatory diseases when variants in both genes are present, this has not been reported for the combination of variants identified in our patient (Yao et al. 2021).
NOD2 is also responsible for Blau Syndrome and Early Onset Sarcoidosis, the hereditary and sporadic forms (respectively) of a rare autoinflammatory disease caused by gain-of-function mutations in NOD2. Affected individuals present with non-caseating granulomatous arthritis, uveitis, and dermatitis (Tangye et al. 2022). Our patient’s symptoms are consistent with Blau syndrome, and in combination with his underlying MEFV mutations, may help to explain his complex presentation. Notably, the c.2104C>T (p.Arg702Trp) variant in NOD2 has not yet been reported in individuals with Blau Syndrome. Still, some features are not completely explained by his genetic work-up, such as malignancy, and further studies may be needed to rule out an association.
In summary, we describe a patient with a complex, longstanding, and early onset autoinflammatory disease. Genetic analysis identified 3 variants of uncertain significance in the MEFV and NOD2 genes.
REFERENCES
Aksentijevich I., Torosyan Y., Samuels J., Centola M., Pras E., Chae J.J., Oddoux C., Wood G., Azzaro M.P., Palumbo G., Giustolisi R., Pras M., Ostrer H., and Kastner D.L. 1999. Mutation and haplotype studies of familial Mediterranean fever reveal new ancestral relationships and evidence for a high carrier frequency with reduced penetrance in the Ashkenazi Jewish population. Am. J. Hum. Genet. 64: 949–962.
Berkun Y., Karban A., Padeh S., Pras E., Shinar Y., Lidar M., Livneh A., and Bujanover Y. 2012. NOD2/CARD15 Gene Mutations in Patients with Familial Mediterranean Fever. Semin Arthritis Rheum. 42: 84–88.
Booty M.G., Chae J.J., Masters S.L., Remmers E.F., Barham B., Le J.M., Barron K.S., Holland S.M., Kastner D.L., and Aksentijevich I. 2009. Familial mediterranean fever with a singleMEFVmutation: Where is the second hit? Arthritis Rheumatol. 60: 1851–1861.
Caruso R., Warner N., Inohara N., and Núñez G. 2014. NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity, 41: 898–908.
Cazeneuve C., Sarkisian T., Pecheux C., Dervichian M., Nedelec B., Reinert P., Ayvazyan A., Kouyoumdjian J.C., Ajrapetyan H., Delpech M., Goossens M., Dode C., Grateau G., and Amselem S. 1999. MEFV-Gene analysis in armenian patients with Familial Mediterranean fever: diagnostic value and unfavorable renal prognosis of the M694V homozygous genotype-genetic and therapeutic implications. Am J Hum Genet 65: 88–97.
Chae J.J., Cho Y.-H., Lee G.-S., Cheng J., Liu P.P., Feigenbaum L., Katz S.I., and Kastner D.L. 2011. Gain-of-Function Pyrin Mutations Induce NLRP3 Protein-Independent Interleukin-1β Activation and Severe Autoinflammation in Mice. Immunity, 34: 755–768.
Chapman J.M., Onnie C.M., Prescott N.J., Fisher S.A., Mansfield J.C., Mathew C.G., Lewis C.M., Verzilli C.J., and Whittaker J.C. 2009. Searching for genotype-phenotype structure: using hierarchical log-linear models in Crohn disease. Am. J. Hum. Genet. 84: 178–187.
Christodoulou K., Wiskin A.E., Gibson J., Tapper W., Willis C., Afzal N.A., Upstill-Goddard R., Holloway J.W., Simpson M.A., Beattie R.M., Collins A., and Ennis S. 2013. Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut, 62: 977–984.
Hannan L.M., Ward J., Ebringer R., and Mcdonald C.F. 2012. Late presentation of familial Mediterranean fever associated with P369S/R408Q variant in the MEFV gene. Intern. Med. J. 42: 952–954.
Hugot J.-P., Chamaillard M., Zouali H., Lesage S., Cézard J.-P., Belaiche J., Almer S., Tysk C., O’Morain C.A., Gassull M., Binder V., Finkel Y., Cortot A., Modigliani R., Laurent-Puig P., Gower-Rousseau C., Macry J., Colombel J.-F., Sahbatou M., and Thomas G. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature, 411: 599–603.
Kacar M., Pathak S., and Savic S. 2019. Hereditary systemic autoinflammatory diseases and Schnitzler’s syndrome. Rheumatology, 58: vi31–vi43.
King K., Sheikh M.F., Cuthbert A.P., Fisher S.A., Onnie C.M., Mirza M.M., Pattni R.C., Sanderson J., Forbes A., Mansfield J., Lewis C.M., Roberts R.G., and Mathew C.G. 2006. Mutation, selection, and evolution of the Crohn disease susceptibility geneCARD15. Human Mutation, 27: 44–54.
Martorana, D., Bonatti, F., Mozzoni, P., Vaglio, A., and Percesepe, A. 2017. Monogenic Autoinflammatory Diseases with Mendelian Inheritance: Genes, Mutations, and Genotype/Phenotype Correlations. Front. Immunol. 8.
Mcgonagle D. and Mcdermott M.F. 2006. A Proposed Classification of the Immunological Diseases. PLoS Med, 3: e297.
Miceli-Richard C., Lesage S., Rybojad M., Prieur A.-M., Manouvrier-Hanu S., Häfner R., Chamaillard M., Zouali H., Thomas G., and Hugot J.-P. 2001. CARD15 mutations in Blau syndrome. Nat. Gen. 29: 19–20.
Migita K., Agematsu K., Yazaki M., Nonaka F., Nakamura A., Toma T., Kishida D., Uehara R., Nakamura Y., Jiuchi Y., Masumoto J., Furukawa H., Ida H., Terai C., Nakashima Y., Kawakami A., Nakamura T., Eguchi K., Yasunami M., and Yachie A. 2014. Familial Mediterranean fever: genotype-phenotype correlations in Japanese patients. Medicine (Baltimore), 93: 158–164.
Mukherjee T., Hovingh E.S., Foerster E.G., Abdel-Nour M., Philpott D.J., and Girardin S.E. 2019. NOD1 and NOD2 in inflammation, immunity and disease. Arc. Biochem. Biophys. 670: 69–81.
Negroni A., Pierdomenico M., Cucchiara S., and Stronati L. 2018. NOD2 and inflammation: current insights. J. Inflam/ Res. 11: 49–60.
Nigrovic P.A., Lee P.Y., and Hoffman H.M. 2020. Monogenic autoinflammatory disorders: Conceptual overview, phenotype, and clinical approach. J. Aller. Clin. Immunol. 146: 925–937.
Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R.H., Achkar J.-P., Brant S.R., Bayless T.M., Kirschner B.S., Hanauer S.B., Nuñez G., and Cho J.H. 2001. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature, 411: 603–606.
Pathak S., Mcdermott M.F., and Savic S. 2017. Autoinflammatory diseases: update on classification diagnosis and management. J. Clin. Pathol. 70: 1–8.
Rood J.E. and Behrens E.M. 2022. Inherited Autoinflammatory Syndromes. Ann. Rev. Pathol. Mech. Disease, 17: 227–249.
Ryan J.G., Masters S.L., Booty M.G., Habal N., Alexander J.D., Barham B.K., Remmers E.F., Barron K.S., Kastner D.L., and Aksentijevich I. 2010. Clinical features and functional significance of the P369S/R408Q variant in pyrin, the familial Mediterranean fever protein. Ann. Rheum. Dis. 69: 1360–1363.
Simon A., Van Der Meer J.W.M., Veselý R., Myrdal U., Yoshimura K., Duys P., and Drenth J.P.H. 2006. Approach to genetic analysis in the diagnosis of hereditary autoinflammatory syndromes. Rheumatology, 45: 269–273.
Steiner A., Harapas C.R., Masters S.L., and Davidson S. 2018. An Update on Autoinflammatory Diseases: Relopathies. Curr. Rheumatol. Rep. 20(7): 39.
Tangye S.G., Al-Herz W., Bousfiha A., Cunningham-Rundles C., Franco J.L., Holland S.M., Klein C., Morio T., Oksenhendler E., Picard C., Puel A., Puck J., Seppänen M.R.J., Somech R., Su H.C., Sullivan K.E., Torgerson T.R., and Meyts I. 2022. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 42(7): 1473–1507.
Tunca M. and Ozdogan H. 2005. Molecular and Genetic Characteristics of Hereditary Autoinflammatory Diseases. Curr. Drug Target –Inflamm. Aller. 4: 77–80.
Yao Q., Li E., and Shen B. 2019. Autoinflammatory disease with focus on NOD2-associated disease in the era of genomic medicine. Autoimmunity, 52: 48–56.
Yao Q. and Shen B. 2017. A systematic analysis of treatment and outcomes of NOD2-associated autoinflammatory disease. Am. J. Med. 130: 365.e13–365.e18.
Yao Q., Shen M., and Gorevic P. 2021. NOD2 versus MEFV: Differential diagnosis of Yao syndrome and familial Mediterranean fever. Rheumatol. Immunol. Res. 2: 233–239.
Yao Q., Zhou L., Cusumano P., Bose N., Piliang M., Jayakar B., Su L.C., and Shen B. 2011. A new category of autoinflammatory disease associated with NOD2 gene mutations. Arthritis Res. Ther. 13: R148.
Yazdanyar S., Weischer M., and Nordestgaard B.G. 2009. Genotyping for NOD2 genetic variants and crohn disease: a metaanalysis. Clin. Chem. 55: 1950–1957.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 9 • Number 4 • December 2022
Pages: 77 - 84
History
Received: 15 November 2022
Accepted: 24 November 2022
Accepted manuscript online: 28 November 2022
Copyright
© 2022.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Laura EdithAbrego Fuentes, IreneChair, PeterVadas, and Chaim M.Roifman. 2022. Heterogeneous phenotype of autoinflammatory disease in a patient with mutations in NOD2 and MEFV genes. LymphoSign Journal.
9(4): 77-84. https://doi.org/10.14785/lymphosign-2022-0013
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
