Association between NOD2 and autoinflammation presenting as Yellow Nail Syndrome
Abstract
Background: Yellow Nail Syndrome is defined as a triad of lymphedema, respiratory symptoms, and nail discolouration. The precise etiology remains unknown, however it has been reported alongside a broad spectrum of conditions including malignancies, autoinflammatory diseases, and immunodeficiencies.
Aim: To highlight the association between defects in the intracellular bacterial sensor gene NOD2 and Yellow Nail Syndrome.
Methods: A retrospective review of the patient’s chart was performed, including family history, characteristics, immune laboratory evaluation, and genetics.
Results: A 65-year-old female was referred to our centre for lymphedema and bronchiectasis. She had recurrent episodes of pneumonia, cellulitis, and oral ulcers. Bilateral lymphedema on her lower limbs up to the hip and discoloured yellow nails were reported. Given her clinical picture, she was diagnosed with Yellow Nail Syndrome. The immunological evaluation was unremarkable overall, with normal T cell subsets and function and adequate antibody titers. Genetic testing identified a heterozygous mutation in the NOD2 gene, c.2107C>T (p.Arg703Cys), considered a variant of uncertain significance.
Conclusion: Heterozygous variants in NOD2 can result in a spectrum of autoimmune and autoinflammatory disorders, including Yellow Nail Syndrome.
Statement of novelty: We describe a patient with Yellow Nail Syndrome, presenting with the classic triad of clinical features. Genetic evaluation identified a heterozygous variant in NOD2, which has been extensively associated with several autoinflammatory diseases, but not Yellow Nail Syndrome.
Introduction
Yellow Nail Syndrome (YNS) was initially described in 1964, consisting of discoloured nails and lymphedema in a group of 13 patients (Samman and White 1964). The clinical features have since evolved to include the triad of discoloured nails, lymphedema, and respiratory symptoms (including pleural effusion, bronchiectasis, sinusitis, and chronic cough) (Hiller et al. 1972; Emerson 1966). At least two of these three manifestations must be present, with nail abnormalities being a major criterion, as the manifestations may not always be present simultaneously (Vignes and Baran 2017). It is reported almost exclusively in adults over 50 years of age, without sex predilection (Hoque et al. 2007; Maldonado et al. 2008; Piraccini et al. 2014). While extremely rare, YNS has also been documented in pediatric populations (Vignes and Baran 2017).
To date, the precise etiology of YNS remains unclear. It has been associated with malignancy (including bronchial carcinoma, breast, non-Hodgkin’s lymphoma, gallbladder, larynx, renal cell carcinoma, endometrium, melanoma, multiple myeloma after hematopoietic stem cell transplantation or precancerous mycosis fungoides), leading to suggestions that YNS is a paraneoplastic syndrome (Hoque et al. 2007; Maldonado et al. 2008; Piraccini et al. 2014; Vignes and Baran 2017). The symptoms of lymphedema, nail discolouration, and pleural effusions (specifically chylothorax) have also been ascribed to lymphatic impairment, yet this does not explain the chronic cough and sinus infections. Microvasculopathy with protein leakage rather than functional lymphatic impairment has also been considered (Maldonado et al. 2008).
An underlying immunological association has previously been proposed (Siegelman et al. 1969). Reports of YNS in conjunction with immunodeficiencies such as common variable immunodeficiency and combined T and B cell deficiency may explain the sinus and pulmonary infections (Vignes and Baran 2017). Yet, immune investigations among these patients are scarce, and the few reported cases are remarkable for severe lymphopenia, deficiency of naive CD4+ and CD8+ T cells and total B cells. Increased transitional B cells were observed, as well as abnormal T cell response to mitogens and antibody deficiency (Gupta et al. 2012).
Some studies have highlighted FOXC2, a member of the forkhead domain family of transcription factors, as a possible genetic cause. Variable phenotypical presentations were described, including lymphedema-distichiasis syndrome (OMIM 153400), Meige lymphedema (OMIM 153200), and lymphedema ptosis (OMIM 15300) (Finegold et al. 2001; Brice 2002).
Here, we describe a patient with YNS who presented with the classic triad of clinical features. Genetic investigations identified a heterozygous mutation in the NOD2 gene, which has been extensively associated with autoinflammatory disease and immunodeficiency.
Case presentation
Patient
Our patient is a 65-year-old female of Italian descent who first presented at the age of 59 years with recurrent X-ray-confirmed pneumonias, occurring 2–3 times per year, which required management with oral antibiotics. She has mild bronchiectasis affecting the right upper and lower lobe, as well as the left lower lobe. Chronic sinusitis was noted, with sinus CT showing mild polypoid hypertrophy of the right inferior nasal turbinate and significant mucosal thickening affecting most of the sinuses. She experiences postnasal drip and chronic cough managed by a corticosteroid inhaler.
Over the past 7 years, she has had 3 separate episodes of cellulitis affecting her left leg, right leg, and once involving her right shin (each treated with IV or oral antibiotics). From an autoimmune standpoint, she suffers recurrent oral ulcers and has osteoarthritis of the hips, knees, and proximal interphalangeal and distal interphalangeal joints. She also has Raynaud’s phenomenon in both hands, triggered by cold weather. An abdominal CT scan demonstrated extensive colon diverticulosis.
On physical examination, the patient was found to have thick yellow nails, inflammatory hyperpigmentation and lichenification, severe lymphedema and lipedema bilaterally on both her lower limbs up to her hips. Given the characteristic symptoms, she was subsequently diagnosed with YNS.
The patient’s family history is remarkable for consanguinity, autoimmunity, and malignancy. Her parents are third cousins. Father suffers from congestive heart disease and hypertension, while the mother has hypothyroidism, hypertension, and arthritis. The patient’s sister passed away from pancreatic cancer around age 60. She has another half-sister and a brother who has hypertension and a history of bladder cancer. The patient has two sons, one had thyroid cancer, and the other suffered from chronic urticaria. Extensive oncology workup of our patient, including whole body CT and mammography, did not show any evidence of malignancy.
Immune evaluation
Immune investigations showed normal immunoglobulins (IgG: 13.6 g/L, IgA 1.9 g/L, IgM: 0.5 g/L) while specific antibody titers against mumps, measles, rubella, tetanus, and varicella were protective (Table 1). Lymphocyte immunophenotyping was unremarkable overall, although slightly low B cell numbers of 83 cells/μL (normal: 91–610 cells/μL) were detected, which is not expected to be significant given normal humoral investigations. T cell subsets were within normal range (CD3+CD8+: 283 cells/μL, normal: 200–900 cells/μL; CD3+CD4+ 1280 cells/μL, normal: 300–1400 cells/μL) with a slight predominance of CD4+ over CD8+ T cells, at a ratio of 4.5 (normal: 0.8–3.9). NK cell numbers were unremarkable. Lymphocyte proliferation to the mitogen phytohemagglutinin (PHA) was normal, with a stimulation index of 1039.
Table 1:
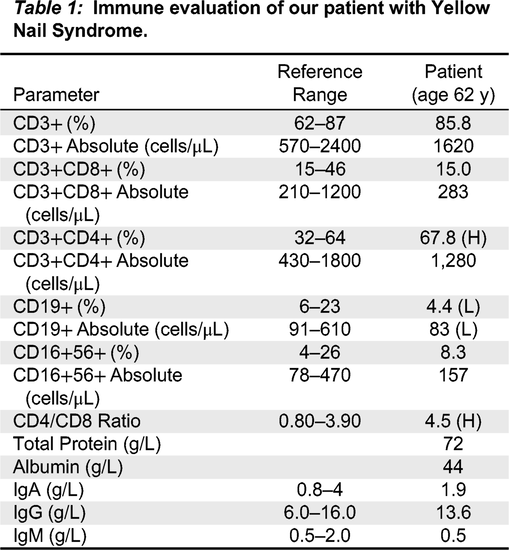
Genetic investigations
A 17-gene Autoinflammatory Disease panel detected a heterozygous variant of uncertain significance in the NOD2 gene (NM_022162.1), c.2107C>T (p.Arg703Cys), resulting in the substitution of an arginine residue with cystine at position 703. In silico programs (Sift, Polyphen, MutationTaster) found no consensus regarding the pathogenicity of this variant.
Discussion
We report a novel association between YNS and the NOD2 gene, encoding NOD2 (nucleotide-binding oligomerization domain-containing protein 2). Previously referred to as CARD15 (caspase recruitment domain-containing protein 15), NOD2 belongs to the intracellular NOD-like receptor family of pattern recognition receptors. These serve as intracellular sensors that detect patterns in bacterial peptidoglycan (i.e., muramyl dipeptide) and promote bacterial clearance through activation of the NF-kappa-B pathway and autophagy. Mutations in NOD2 are linked with several chronic inflammatory diseases, suggesting that balanced signalling is critical for maintaining immune homeostasis (Negroni et al. 2018). NOD2 is expressed primarily in monocytes but also in macrophages, dendritic cells, lymphocytes, epithelial and endothelial cells, and intestinal paneth cells.
The NOD2 gene maps to chromosome 16q12 and comprises 12 coding exons. Structurally, NOD2 consists of 2 N-terminal Caspase Recruitment Domains (CARD), followed by a Nucleotide Binding Domain (NBD, residues 195–425), Helicoidal Domain 1 (HD1, residues 426–485), Winged-helix Domain (WHD, residues 486–602), Helicoidal Domain 2 (HD2, residues 2603–743) and a C-terminal leucine-rich repeat domain (LRR, residues 744–1,020) (Maekawa et al. 2016) (Figure 1). The CARD domains are responsible for signal transduction, the NBD through HD2 domains together form the NOD domain with ATPase activity, while the LRR is essential for recognizing ligands.
Figure 1:
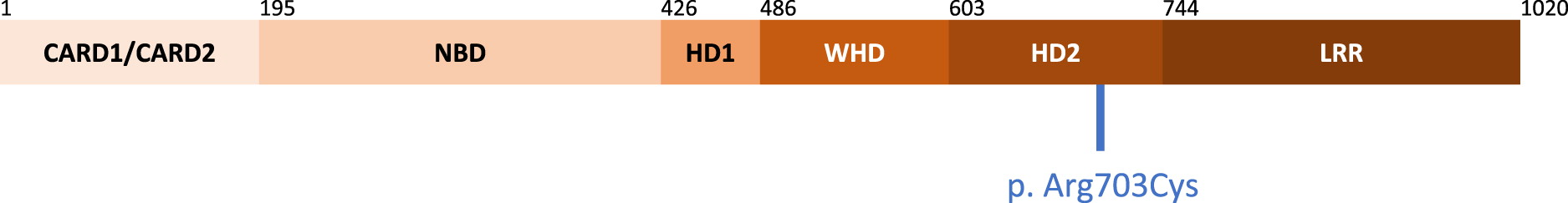
To date, pathogenic mutations in NOD2 have been associated with Blau Syndrome (OMIM 186580) (Miceli-Richard et al. 2001) and Early Onset Sarcoidosis, Crohn’s Disease (OMIM 266600) (Ogura et al. 2001; Hugot et al. 2001; King et al. 2006), and NOD Associated Autoinflammatory Disease (NAID)/Yao Syndrome (OMIM 617321) (Yao et al. 2011). In our patient, the heterozygous NOD2 variant, c.2107C>T (p.Arg703Cys), affects the HD2 domain. Mutations targeting this domain have been reported in patients with the above conditions, resulting in either gain- or loss-of-function (Tanabe et al. 2004).
Blau Syndrome and Early Onset Sarcoidosis are the heritable and sporadic forms, respectively, of an autoinflammatory disease caused by NOD2 gain-of-function mutations. These are characterized by non-caseating granulomatous arthritis, uveitis, and dermatitis. Cranial neuropathies, sarcoidosis, and a high incidence of Crohn’s colitis are reported (Tangye et al. 2022). The majority of NOD2 mutations that lead to these conditions are concentrated on or near the domain interfaces of NBD, HD1, WHD and HD2: 3 mutations have been reported in the NBD, 7 in HD1, 3 in the WHD, and 1 in HD2. While arthritis involving wrists, ankles, knees, and interphalangeal joints in Blau syndrome/Early Onset is reminiscent of the arthritic features in our patient, the other more common features were absent.
The NOD2 variants associated with Crohn’s disease, of which the majority are loss-of-function, can be found scattered throughout the NOD2 protein in all domains except HD1; 13 mutations mapped to the NBD, 1 to the WHD, 7 to HD2 and 10 to the LRR domain (Maekawa et al. 2016). Of note, a polymorphism associated with Crohn’s disease, p.Arg702Trp, lies within just one residue of the mutation identified in our patient. Yet, the only gastrointestinal finding in our patient was diverticulosis.
The p.Arg702Trp is also present in patients described with NAID/Yao Syndrome, an autoinflammatory disease characterized by periodic fever, dermatitis, arthritis, and swelling of the distal extremities, as well as gastrointestinal and sicca-like symptoms (Yao et al. 2013, 2019). The clinical picture of our patient does not fit the NAID/Yao Syndrome criteria.
The features of YNS in our patient may be an atypical or mild presentation of one or more of the diverse conditions attributed to NOD2. Paradoxically, her immune evaluation was not consistent with the previously described immune defects associated with YNS. It is not uncommon for genes located on the same chromosome to share overlapping phenotypes. For example, inflammatory disorders categorized under the umbrella of Systemic Autoinflammatory Diseases (SAIDs), which encompasses a group of genetically diverse and heterogeneous inflammatory disorders resulting from abnormal innate immunity, have been attributed to both NOD2 and MEFV genes (Aksentijevich 2015; Yao et al. 2019). Both are present in chromosome 16. Interestingly, FOXC2, linked to YNS (OMIM 602402), is also located on chromosome 16.
In sum, we describe a patient with YNS whom, upon genetic evaluation, we identified a heterozygous mutation in NOD2. To our knowledge, this is the first report of such an association. Further investigations are required to delineate the role of NOD2 in YNS. With the widespread availability of genetic tools, including next-generation sequencing, attaining a genetic diagnosis should be prioritized to provide tailored and effective treatment options.
REFERENCES
Aksentijevich I. 2015. Update on genetics and pathogenesis of autoinflammatory diseases: the last 2 years. Semin. Immunopathol. 37: 395–401.
Brice G. 2002. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J. Med. Genet. 39: 478–483.
Emerson P.A. 1966. Yellow nails, lymphoedema, and pleural effusions. Thorax, 21: 247–253.
Finegold D.N., Kimak M.A., Lawrence E.C., Levinson K.L., Cherniske E.M., Pober B.R., Dunlap J.W., and Ferrell R.E. 2001. Truncating mutations in FOXC2 cause multiple lymphedema syndromes. Hum. Mol. Genet. 10: 1185–1189.
Gupta S., Samra D., Yel L., and Agrawal S. 2012. T and B Cell Deficiency Associated with Yellow Nail Syndrome. Scand. J. Immunol. 75: 329–335.
Hiller E., Rosenow E.C., and Olsen A.M. 1972. Pulmonary Manifestations of the Yellow Nail Syndrome. Chest, 61: 452–458.
Hoque S.R., Mansour S., and Mortimer P.S. 2007. Yellow nail syndrome: not a genetic disorder? Eleven new cases and a review of the literature. Br. J. Dermatol. 156: 1230–1234.
Hugot J.P., Chamaillard M., Zouali H., Lesage S., Cezard J.P., Belaiche J., Almer S., Tysk C., O’morain C.A., Gassull M., Binder V., Finkel Y., Cortot A., Modigliani R., Laurent-Puig P., Gower-Rousseau C., Macry J., Colombel J.F., Sahbatou M., and Thomas G. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature, 411: 599–603.
King K., Sheikh M.F., Cuthbert A.P., Fisher S.A., Onnie C.M., Mirza M.M., Pattni R.C., Sanderson J., Forbes A., Mansfield J., Lewis C.M., Roberts R.G., and Mathew C.G. 2006. Mutation, selection, and evolution of the Crohn disease susceptibility gene CARD15. Hum. Mutat. 27: 44–54.
Maekawa S., Ohto U., Shibata T., Miyake K., and Shimizu T. 2016. Crystal structure of NOD2 and its implications in human disease. Nat. Commun. 7: 11813.
Maldonado F., Tazelaar H.D., Wang C.-W., and Ryu J.H. 2008. Yellow Nail Syndrome. Chest, 134: 375–381.
Miceli-Richard C., Lesage S., Rybojad M., Prieur A.M., Manouvrier-Hanu S., Hafner R., Chamaillard M., Zouali H., Thomas G., and Hugot J.P. 2001. CARD15 mutations in Blau syndrome. Nat. Genet. 29: 19–20.
Negroni A., Pierdomenico M., Cucchiara S., and Stronati L. 2018. NOD2 and inflammation: current insights. J. Inflamm. Res., 11: 49–60.
Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R.H., Achkar J.P., Brant S.R., Bayless T.M., Kirschner B.S., Hanauer S.B., Nunez G., and Cho J.H. 2001. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature, 411: 603–606.
Piraccini B.M., Urciuoli B., Starace M., Tosti A., and Balestri R. 2014. Yellow nail syndrome: Clinical experience in a series of 21 patients. JDDG: J. Dtsch. Dermatol. Ges. 12: 131–137.
Samman P.D. and White W.F. 1964. The “Yellow Nail” Syndrome. Br. J. Dermatol. 76: 153–157.
Siegelman S.S., Heckman B.H., and Hasson J. 1969. Lymphedema, Pleural Effusions and Yellow Nails. Dis. Chest. 56: 114–117.
Tanabe T., Chamaillard M., Ogura Y., ZHU L., Qiu S., Masumoto J., Ghosh P., Moran A., Predergast M.M., Tromp G., Williams C.J., Inohara N., and Núñez G. 2004. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J. 23: 1587–1597.
Tangye S.G., Al-Herz W., Bousfiha A., Cunningham-Rundles C., Franco J.L., Holland S.M., Klein C., Morio T., Oksenhendler E., Picard C., Puel A., Puck J., Seppänen M.R.J., Somech R., Su H.C., Sullivan K.E., Torgerson T.R., and Meyts I. 2022. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 24: 1–35.
Vignes S. and Baran R. 2017. Yellow nail syndrome: a review. Orphanet. J. Rare. Dis. 12(1): 42.
Yao Q., Li E., and Shen B. 2019. Autoinflammatory disease with focus on NOD2-associated disease in the era of genomic medicine. Autoimmunity, 52: 48–56.
Yao Q., Su L.-C., Tomecki K.J., Zhou L., Jayakar B., and Shen B. 2013. Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J. Am. Acad. Dermatol. 68: 624–631.
Yao Q., Zhou L., Cusumano P., Bose N., Piliang M., Jayakar B., Su L.C., and Shen B. 2011. A new category of autoinflammatory disease associated with NOD2 gene mutations. Arthritis. Res. Ther. 13: R148.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 9 • Number 3 • September 2022
Pages: 67 - 71
History
Received: 21 August 2022
Accepted: 28 August 2022
Accepted manuscript online: 30 August 2022
Copyright
© 2022.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Laura EdithAbrego Fuentes and MohammadAlsalamah. 2022. Association between NOD2 and autoinflammation presenting as Yellow Nail Syndrome. LymphoSign Journal.
9(3): 67-71. https://doi.org/10.14785/lymphosign-2022-0011
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
