A clinical trial protocol to evaluate the safety and pharmacokinetics of subcutaneously administered immunoglobulin in patients with primary immunodeficiency
Abstract
This protocol is excerpted from recent clinical trials used to study the pharmacokinetics, safety, and tolerability of subcutaneously administered immunoglobulin (SCIG) in subjects with primary immunodeficiency. The primary objective is to determine the weekly dose of SCIG product that produces a steady-state area under the concentration-time curve of total immunoglobulin G level that is non-inferior to that of regularly administered intravenous immunoglobulin (IVIG). We include details of the target population, eligibility criteria, treatment phases, key assessments and procedures, and study analyses. Given that IVIG may be problematic in patients with poor venous access or those who develop systemic adverse effects, among others, the development of SCIG for use in the home setting provides an alternative treatment technique for adults and children with primary immunodeficiency.
Statement of novelty: This protocol describes the main topics found in prospective clinical studies evaluating the safety and pharmacokinetics of SCIG in subjects with primary immunodeficiency.
Introduction
Primary immunodeficiency
Primary immunodeficiency diseases (PIDs) are a group of rare, chronic disorders caused by either de novo or hereditary mutations in genes of the immune system. Such changes may alter the development or function of individual immune cell types, affect the response to an immune trigger, or broadly affect a common cellular process required for immunological competence. Clinically, patients with PID experience frequent bacterial, fungal, protozoal, and viral infections (Picard et al. 2015). B cell deficiencies, also known as humoral or antibody deficiencies, comprise the largest group of PIDs and are characterized by insufficient or absent levels of protective antibodies. While the underlying cause is often due to mutations within genes regulating B cell differentiation, in many instances, the molecular basis remains unknown. Hypogammaglobulinemia and agammaglobulinemia are common features of X-linked agammaglobulinemia (XLA) (Bruton 1952; Bonilla and Geha 2003) and common variable immunodeficiency (CVID) (Resnick and Cunningham-Rundles 2012).
It is notable that immunoglobulin (Ig) G replacement therapy, first introduced in 1952 with the infusion of serum globulin fraction, has proven efficacious in reducing the frequency of infections among these patients (Roifman and Gelfand 1988). To date, the therapeutic management of PID has been carried out using various IgG preparations via intramuscular (IM), intravenous (IV), and subcutaneous (SC) routes.
Immunoglobulin replacement therapy
Infusion of IV IgG (IVIG) is widespread, however, in a subset of patients with poor venous access or those who develop systemic adverse events (AE), such as headaches, fever, chills, and myalgia (Ochs et al. 2006), delivery of IgG by SC infusion (SCIG) represents an alternative that is safe and has good efficacy (Stiehm et al. 1998; Radinsky and Bonagura 2003). Whereas IVIG is associated with marked variability in IgG levels between infusions (coinciding with a rise in IgG immediately following infusion and a decline after redistribution to the extravascular space), SCIG is absorbed and redistributed slowly, resulting in higher and more consistent trough levels (Orange et al. 2012; Berger and Allen 2015). It is generally accepted that SCIG causes few, if any, adverse systemic reactions and such home-based therapy may afford patients greater independence, freedom from reliance on trained personnel and specialized facilities, as well as improved quality-of-life.
Study rationale
The primary objective is to determine a weekly dose of SCIG product (referred to herein as SCIG-x) that produces a steady-state area under the concentration-time curve (AUC) of total IgG level that is non-inferior to that of a regularly administered IVIG product (referred to as IVIG-x). Given that the bioavailability of SCIG is reportedly 30% lower compared to administration of IVIG, due in part to extracellular matrix binding and breakdown by the tissue enzymes (Wang et al. 2008; Berger et al. 2013), an IVIG to SCIG dose adjustment of 1.37 will be used. This is based on historical data assessing bioequivalence of these routes of infusion (Wasserman et al. 2010, 2011; Berger et al. 2011), and addresses the requirement of the US Food and Drug Administration (US FDA) that doses of SCIG be adjusted to provide equivalent AUC of total IgG compared to that of previous IVIG therapy. The dose adjustment factor of 1.37 will be evaluated in interim pharmacokinetic (PK) analysis and modified if necessary.
Study objective
The objective of this study is to evaluate the pharmacokinetics, safety, and tolerability of an SCIG in subjects with PID.
Primary pharmacokinetic objective
To determine a dose of weekly SCIG-x that produces a steady-state AUC of total IgG by SC administration that is not inferior to that of a regularly administered IV dose of IVIG-x in subjects with PID.
Secondary pharmacokinetic objective
To determine whether SCIG-x maintains steady-state trough total IgG levels that are comparable to the mean trough total IgG levels of IVIG-x in subjects with PID.
Safety objective
To assess the safety and tolerability of the SCIG-x formulation.
Exploratory objectives
To evaluate:
- Maximum concentration (Cmax) and time to reach Cmax (Tmax)
- Trough levels of IgG subclasses and measles antibody subclasses
- Antibody titres of Streptococcus pneumoniae, Hemophilus influenzae, and Clostridium tetani
- Rate of serious bacterial infections
- All types of infections as well as validated infections
- Number of days on antibiotics (prophylactic or for treatment of infection)
- Number of hospitalizations due to infections
- Number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
Investigational plan
The study is a prospective, multi-center, open-label, single-sequence study. To account for possible dropouts, 50 subjects with well-defined PID and antibody deficiency will be enrolled to ensure approximately 30 adult subjects and 10 pediatric subjects (age 2–16 years) complete treatment with SCIG-x. This number of subjects would provide enough data to assess the primary objective. All subjects will be followed for 6 months. Pharmacokinetic (PK) samples will be collected to assess total IgG profiles.
Study duration
The study period is expected to be up to 50 weeks, including 7–28 days for screening. There are 3 treatment phases (Figure 1): run-in phase (3–4 m), IV phase (4–5 weeks), and SC phase (24 weeks). A follow-up visit will be scheduled 21 or 28 days after the last study infusion.
Figure 1:
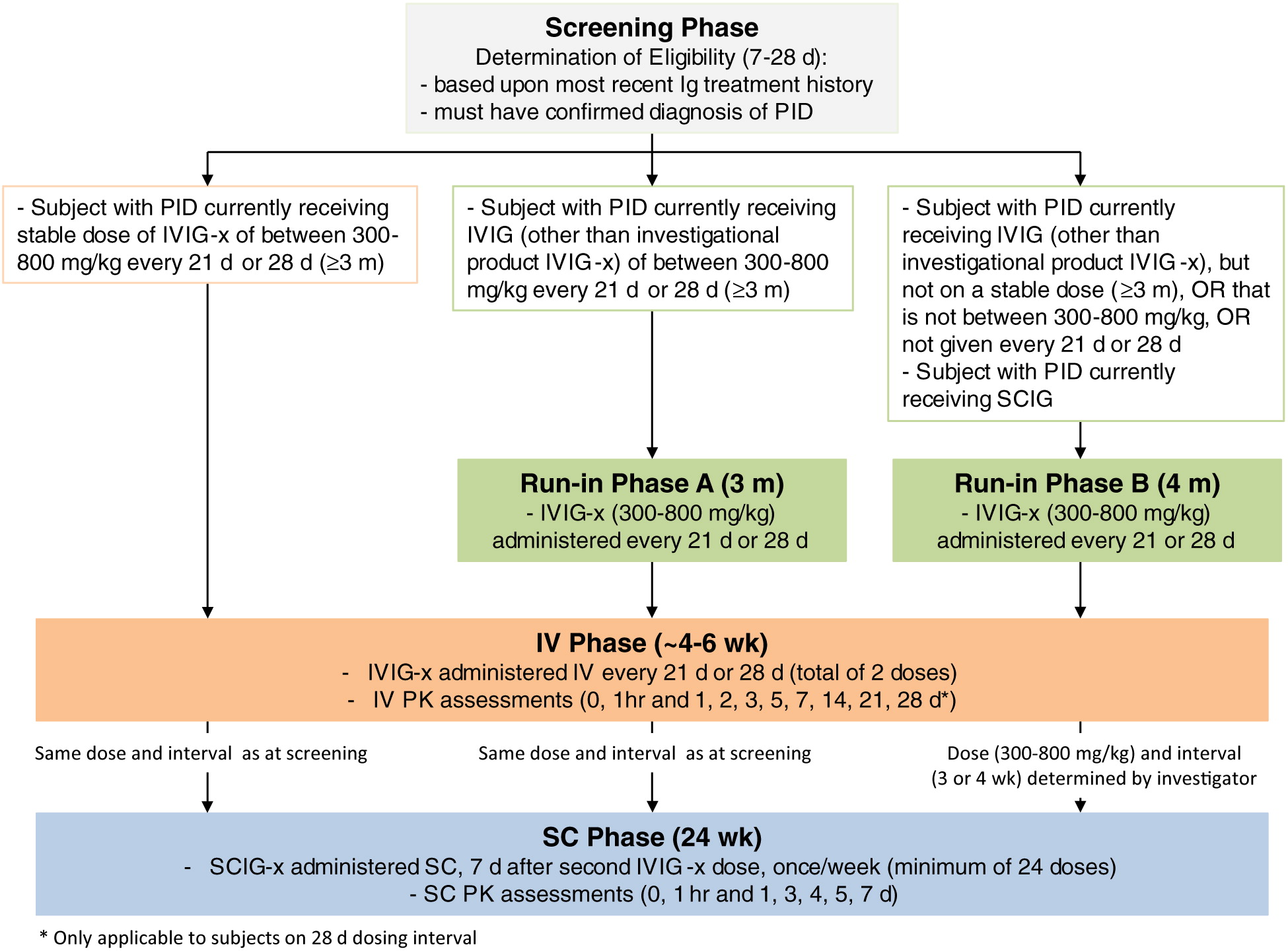
Study design
All subjects will be screened for between 7 and 28 days before enrollment into the study and, if eligible, will enter the run-in or IV phase (IV administration of IVIG-x) based upon the most recent IgG treatment history. Subjects will subsequently receive SC administration of SCIG-x during the SC phase, using a dose adjustment factor of 1.37.
Enrolled subjects who currently receive a stable IV dose (≥3 m) of the study drug IVIG-x (300–800 mg, every 3 or 4 weeks) will bypass the run-in phase and proceed directly to the IV phase. Their first infusion of IVIG-x will coincide with their next scheduled IV infusion, in accordance with their regular dosing interval.
Run-in phase
Subjects will be assigned entry into run-in phase A if they were previously receiving IVIG therapy (300–800 mg/kg, every 3 or 4 weeks) but not IVIG-x (i.e., a different commercially available drug). The subjects in this category will receive IVIG-x, at an equivalent dose and interval prior to study entry, for a total duration of 3 m.
Subjects will be assigned entry into run-in phase B if they were previously receiving either SCIG or IVIG therapy, but not at a stable IV dose for 3 m prior to screening, or where the dose is not 300–800 mg/kg, or where the infusions were not at an interval of 3 or 4 weeks. The subjects in this category will receive IVIG-x, at an equivalent dose and interval prior to study entry, for a total duration of 4 m.
IV phase
All subjects will receive 2 in-clinic IV doses of IVIG-x at their regular dosing interval (every 3 or 4 weeks). PK samples will be collected pre-dose and post-dose (0 and 1 hours, and 1, 2, 3, 5, 7, 14, and 21 days. Subjects on a 4 week interval will also have samples collected at 28 days).
SC phase
Seven days after the second IV infusion of IVIG-x, subjects will begin weekly SC infusions of the investigational product SCIG-x. Weekly infusions will take place for 6 m, with PK measurements performed after 3 m (SC infusion #13; 0 and 1 hours, and 1, 3, 4, 5, 7 days). The SC dose will be calculated using the IV to SC dose adjustment factor of 1.37:
Ten infusions will take place in-clinic (SC infusion #1, 2, 3, 5, 9, 13, 14, 17, 21). All others will occur in the home setting. A final in-clinic visit will take place at week 25.
Interim PK analysis
Interim PK analysis will be performed to evaluate whether the dose adjustment factor is adequate to provide steady-state AUC of total IgG levels that is not inferior to levels achieved during the prior IV dose. Analysis will be based on the first 6 subjects (age ≥12–75 years) to complete PK assessments during both the IV phase and SC phase (i.e., after SC infusion #14). Standard PK analysis methods will be used: if the geometric least-squares mean (LSM) ratio of SC AUC of total IgG vs. IV AUC of total IgG ≥0.9, no changes will be made to the dose adjustment factor. If the ratio is <0.9 and mean trough concentrations of IgG falls below the target level during administration of SCIG-x, the dose adjustment factor will be increased with consideration given to mean IgG trough levels obtained during SCIG-x administration. A revised dose adjustment factor will be communicated to sites and implemented at the next in-clinic infusion. All subjects will be required to complete 24 weeks of SC infusions with the revised dose adjustment factor, with the first 6 subjects undergoing PK analysis after 12 weeks (as above).
Investigators and study centres
Study centres will enroll up to 50 subjects.
Randomization and stratification
Not applicable.
Blinding
Not applicable.
Selection of the study population
Subjects with a confirmed diagnosis of PID requiring IgG replacement will be selected for screening. Those who fail to meet eligibility criteria may be re-screened once.
Inclusion criteria
1.
Male or female, between ages of 2–75 years (inclusive) at the time of screening.
2.
Confirmed clinical diagnosis of PID (IUIS classification) with agammaglobulinemia or hypogammaglobulinemia requiring IgG replacement therapy.
3.
Subject has not had documented serious bacterial infection (SBI) within the 3 months prior to screening.
4.
Subject has received licensed IgG replacement therapy (IV or SC) for at least 3 consecutive months prior to this study. Those on IVIG must currently receive a dosage of 300–800 mg/kg per infusion.
5.
At least 1 documented IgG trough level of ≥500 mg/dL within the last 3 months while on current IgG replacement therapy.
6.
Screening trough IgG level must be ≥500 mg/dL. If below this threshold, the subject will be logged as a screen failure. The subject may be screened a second time following dose adjustment and maintenance of stable dosing for a period of at least 3 consecutive months.
7.
Authorization to access personal health information, including medical records.
8.
Subject, parent, or guardian has signed an informed consent. If applicable, pediatric subjects will require assent forms as appropriate per study documentation and approved by the local jurisdiction (Research Ethics Board/Institutional Review Board).
9.
Subject/caregiver is willing to comply with all aspects of the protocol.
Exclusion criteria
1.
Subject has previously had a serious adverse reaction to Ig or severe anaphylactic reaction to blood or any blood-derived product.
2.
Subject has a history of disorders where SC therapy may be contraindicated, including blistering skin disease, clinically significant thrombocytopenia, bleeding disorder, recurrent skin infections, or diffuse rash.
3.
Subject has been diagnosed with isolated IgG subclass deficiency, isolated specific antibody deficiency, or transient hypogammaglobulinemia of infancy.
4.
Subject has isolated IgA deficiency (with or without antibodies to IgA).
5.
Females who are pregnant, breast feeding, or planning a pregnancy during the course of the study. Subjects who become pregnant during the study will be withdrawn from the study.
6.
Subject has significant proteinuria (dipstick proteinuria is ≥3, urinary protein loss >1–2 g/24 hours, nephrotic syndrome), protein losing enteropathy, history of acute renal failure, severe renal impairment (blood urea nitrogen or creatinine >2.5 times the upper normal limit), receiving dialysis, nephrotic syndrome, or lymphangiectasia.
7.
Subject has levels of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) >2.5 times the upper limit of normal range for the testing laboratory.
8.
Subject has hemoglobin levels <9 g/dL.
9.
Subject has a history (incident in the year prior to screening, or 2 episodes during lifetime) or current diagnosis of thromboembolism or deep venous thrombosis, or any other significant acute or chronic disease that, in the opinion of the investigator, may place the subject at undue medical risk or interfere with the subjects’ ability to complete the study.
10.
Subject is currently receiving anti-coagulant therapy which would make SC route of administration inadvisable.
11.
Subject currently has a known hyperviscosity syndrome.
12.
Subject has secondary immunodeficiency or human immunodeficiency virus (HIV) infections/acquired immune deficiency syndrome (AIDS).
13.
Subject has a known history, or is positive at enrolment, for current hepatitis B virus (HBV) or hepatitis C virus (HCV) infection.
14.
Subject (≤18 years of age) has non-controlled arterial hypertension ≥90th percentile systolic or diastolic blood pressure for age and height; or, if subject is an adult, has non-controlled arterial hypertension (systolic blood pressure >160 mm Hg and (or) diastolic blood pressure >100 mm Hg).
15.
Subject is receiving the following medication: (i) long-term systemic steroids (dose >1 mg/kg/day of prednisone or equivalent for >30 days); (ii) immunosuppressive drugs or chemotherapy; and (iii) immunomodulators.
16.
Subject has known prescription drug or substance abuse.
17.
Subject has participated in another clinical study (interventional) within 3 weeks prior to study enrollment.
Withdrawal of subjects
Subjects can withdraw from the study at their own request, or at the request of their legally acceptable representative, without prejudice. In all cases, the reason for withdrawal will be recorded. The investigator may withdraw a subject from the study drug or the study for the following reasons:
- If, in the investigator’s opinion, continuation in the study poses a risk or would be detrimental to the well-being of the subject (for example, following occurrence of severe or serious AE, or medical condition)
- Subject develops an SBI during the IV phase of the study, prior to the first dose of SCIG-x
- Subject does not comply with the protocol
- Pregnancy
- At the specific request of the sponsor
Premature termination of study/closure of centre
During the study, the Sponsor, REB/IRB and (or) regulatory authorities have the right to terminate the study or close the study centre at any time. The REB/IRB must be informed. In the event of premature study termination or site closure, all materials must be returned to the sponsor with exception of documents that have to remain stored on site. A study centre may be closed due to:
- Insufficient enrolment
- Non-compliance with the study protocol
- Unexpected and significant or unacceptable risks for study subjects, usually due to non-compliance with ICH guideline for Good Clinical Practice (GCP)
Study conduct
Treatments regimens
Subjects will be assigned entry into run-in phase A, run-in phase B, or IV phase, based on the subject’s current antibody replacement regimen (refer to Study Design, Figure 1). Subjects entering run-in phase A or run-in phase B will be administered 300–800 mg/kg IVIG-x (every 21 or 28 days, depending on current dose interval) for 3 or 4 m, respectively. Subjects entering directly into the IV phase will be administered 2 doses of IVIG-x (300–800 mg/kg of body weight every 21 or 28 days). All subjects will proceed to the SC phase of the study, and receive weekly doses SCIG-x according to the IV to SC dose adjustment calculation formula. The dosing of IVIG-x and SCIG-x will be dependent on the treatment each subject receives at the time of screening. This will remain unless there is a medically justified need to change it or if there is a change made to the dose adjustment factor.
Treatment assignment
A subject will be enrolled into the study once written informed consent/assent has been obtained and inclusion/exclusion criteria is fulfilled. Assignment of the treatment phase (run-in phase A, run-in phase B, or IV phase) will be based on the subject’s current dose of antibody replacement product and interval.
Subject identification
Subjects will be assigned a numerical subject identifier in the format xxx-yyyy. This is based on a 3-digit study centre number (xxx, assigned by the sponsor) and a sequential 4-digit subject number designated by the investigator at the screening visit (yyyy). Once a subject identifiers has been assigned it will not be reused at any centre.
Physical examination
A full physical examination will be performed during the screening visit, first SCIG-x infusion (SC infusion 1) and the final visit/termination visit. This will include evaluation of all body systems as considered normal standard of care for the centre. Abbreviated physical examinations will be performed during all other visits, and will target symptoms as well as examination of heart, lung, ears/nose/throat, inspection of previous injection sites.
Medical history and demography
Medical records will be reviewed in detail to confirm documented diagnosis of PID, including current and previous IgG treatments. Demographics including age, gender and history of PID diagnostic tests will be recorded in case report forms (CRF).
Subject diary
Subjects will be provided an infusion diary prior to the first SC infusion and given careful instructions to record items daily. Entries made since the previous clinic visit will be reviewed at every in-clinic visit. The following will be entered into the diary:
- Local infusion site reactions
- Concomitant medications, prescription and non-prescription
- Details of study drug infusion (number and location of sites, date and (or) time of start/end, dose and (or) volume of each dose, duration and rate)
- Number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
- Adverse events (AE) that occurred following the previous study infusion
Dosage
The amount of IVIG-x and SCIG-x to be administered will be calculated based on each subject’s current IgG replacement regimen (refer to Figure 1). For SC infusions, the weekly dose will be one quarter (for subjects on 4 week dosing regimens) or one third (for subjects on 3 week dosing regimens) of the IV dose of IVIG-x, multiplied by the dose adjustment factor of 1.37. The subject’s weight will be recorded at screening and prior to each infusion to determine if adjustment of the dose is necessary.
Subcutaneous administration
The subject and investigator will determine the number of injections sites, rate of infusion, and the time of day that the SC infusions will take place, with emphasis on adhering to the dosing regimen without deviation. The subject will receive the first 3 infusion in clinic under supervision to ensure adequate training before self-administration in the home setting is allowed.
The same anatomical region may be used for SC infusions, and regions may be rotated throughout the study. A maximum of 8 infusion sites will be used per infusion, with no less than 2 in. between sites and target infusion rates not exceeding 25 mL/hour/site. Target infusion rates should not be changed once the rate is reached, unless there are tolerability issues.
Visit schedule
The schedule of study procedures is summarized in Table 1. Each study visit will be allowed a window of ±1 day. All IVIG procedures will take place in the clinic setting. During the first 3 SC phase infusions, subjects will be trained to self-administer the SCIG product in the clinic. Subsequent infusions will be done in the home setting with the exception of infusions 5, 9, 13, 14, 17, and 21.
Table 1:
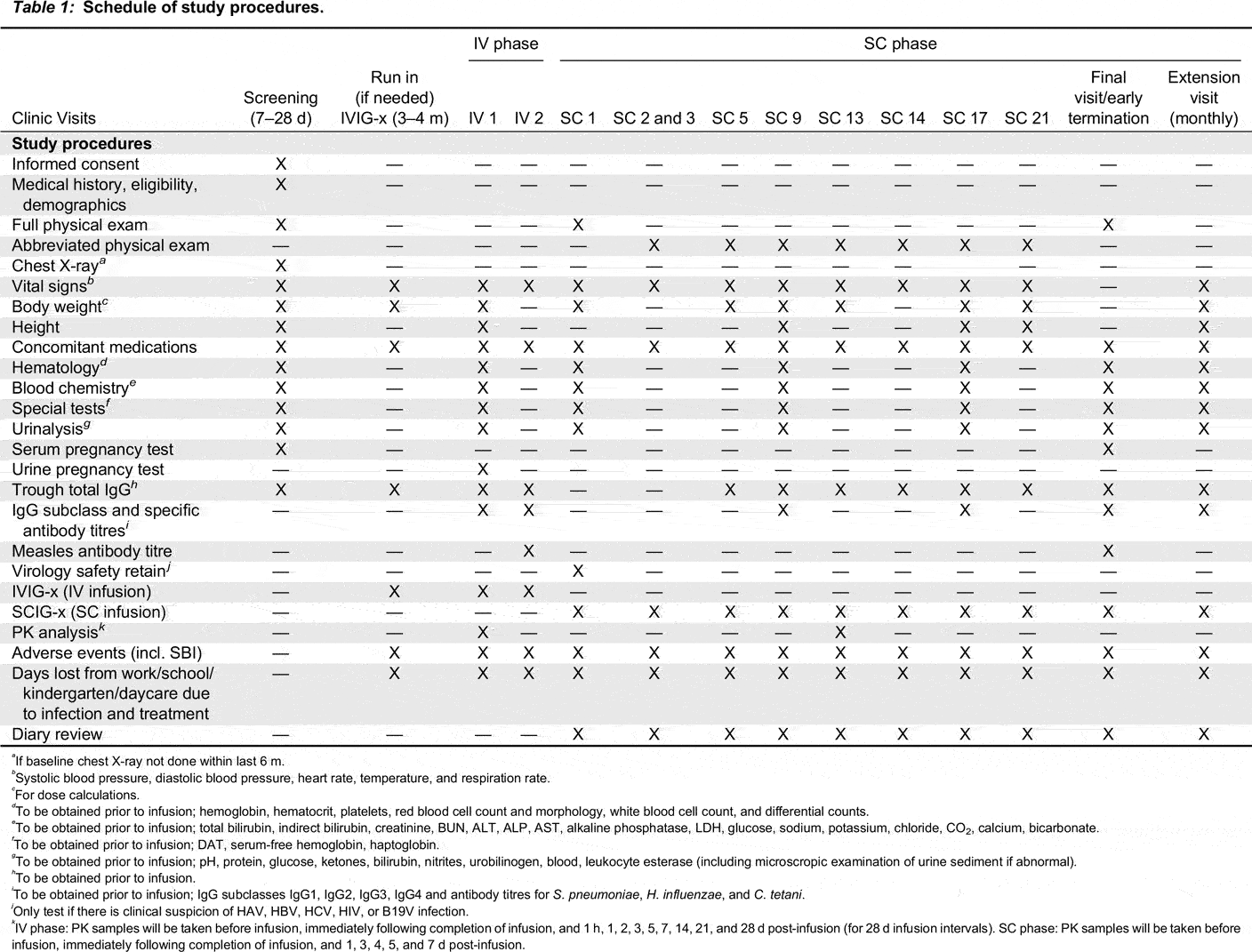
a
If baseline chest X-ray not done within last 6 m.
b
Systolic blood pressure, diastolic blood pressure, heart rate, temperature, and respiration rate.
c
For dose calculations.
d
To be obtained prior to infusion; hemoglobin, hematocrit, platelets, red blood cell count and morphology, white blood cell count, and differential counts.
e
To be obtained prior to infusion; total bilirubin, indirect bilirubin, creatinine, BUN, ALT, ALP, AST, alkaline phosphatase, LDH, glucose, sodium, potassium, chloride, CO2, calcium, bicarbonate.
f
To be obtained prior to infusion; DAT, serum-free hemoglobin, haptoglobin.
g
To be obtained prior to infusion; pH, protein, glucose, ketones, bilirubin, nitrites, urobilinogen, blood, leukocyte esterase (including microscropic examination of urine sediment if abnormal).
h
To be obtained prior to infusion.
i
To be obtained prior to infusion; IgG subclasses IgG1, IgG2, IgG3, IgG4 and antibody titres for S. pneumoniae, H. influenzae, and C. tetani.
j
Only test if there is clinical suspicion of HAV, HBV, HCV, HIV, or B19V infection.
k
IV phase: PK samples will be taken before infusion, immediately following completion of infusion, and 1 h, 1, 2, 3, 5, 7, 14, 21, and 28 d post-infusion (for 28 d infusion intervals). SC phase: PK samples will be taken before infusion, immediately following completion of infusion, and 1, 3, 4, 5, and 7 d post-infusion.
Screening: day 28 or day 21 to day 7
- Obtain written informed consent (and assent if applicable) prior to accessing personal health information
- Confirm eligibility by checking whether subject meets inclusion/exclusion criteria
- Record demography and medical history, including age of PID diagnosis, previous/current IgG therapy
- Full physical examination, body weight, height, vital signs (SBP, systolic blood pressure; DBP, diastolic blood pressure; HR, heart rate; T, temperature; RR, respiration rate)
- Chest X-ray (if not performed within 6 m prior to screening)
- Prior and concomitant medications
- Blood and urine for clinical laboratory assessments:
○ Hematology; including complete blood count, white blood differential and platelet count
○ Blood chemistry; including total bilirubin, creatinine, blood urea nitrogen (BUN), alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, lactate dehydrogenase (LDH), glucose, sodium, potassium, chloride, CO2, calcium
○ Special tests; direct antiglobulin (DAT), serum free hemoglobin, haptoglobin
○ Urinalysis; including microscopic examination of urine sediment
○ Pregnancy test for females of child-bearing potential
- Documented total IgG trough level ≥500 mg/dL
If eligibility is confirmed, subjects will commence their first run-in phase visit or IV phase visit at their next scheduled IgG infusion, in accordance with their regular dosing interval.
Run-in phase (if required)
- Record vital signs
- Body weight
- Record concomitant medications
- Take blood sample for pre-dose trough total IgG
- Administer study product IVIG-x
- Record AE (subjects with SBI will be withdrawn from the study)
- Record number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
IV phase, baseline visit 1 and PK assessment
- Record vital signs
- Body weight (for dose calculation) and height
- Take samples prior to infusion (within 0.5 hours) for hematology, clinical chemistry, special tests, trough total IgG, IgG subclass, antibody titer levels for S. pneumoniae, H. influenzae, and C. tetani
- Urinalysis sample
- Pregnancy test for females of child-bearing potential
- Record concomitant medications
- Administer study product IVIG-x
- Document date and time of start and end of infusion, amount and rate of dose infused
- Record AE
- Record number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
- Take samples for PK assessment of total IgG levels at the following time points:
○ Prior to infusion (within 0.5 hours)
○ Immediately following completion of infusion
○ Post infusion—1 hour (±2 hours), 1 day (±2 hours), 2 days (±2 hours), 3 days (±4 hours), 5 days (±4 hours), 7 days (±1 day), 14 days (±1 day), 21 days (±1 day; final sample for subjects on a 21 days interval and will also be used as pre-dose blood sample for next visit), 28 days (±1 day; only for subjects on a 28 days interval and will also be used as pre-dose blood sample for next visit)
IV phase, visit 2
- Record vital signs
- Concomitant medications
- Take sample prior to infusion for trough total IgG, IgG subclass and antibody titer levels for S. pneumoniae, H. influenzae, and C. tetani (same as post-infusion 21 or 28 days PK sample)
- Blood drawn pre-infusion for measles antibody titer
- Administer study product IVIG-x
- Record AE (subjects with SBI will be withdrawn from the study)
- Record number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
SC phase, infusion week 1
- Full physical examination, body weight (for dose calculation), height, record vital signs
- Concomitant medications
- Blood and urine for clinical laboratory assessments:
○ Hematology; including complete blood count, white blood differential and platelet count
○ Blood chemistry; including total bilirubin, creatinine, BUN, ALT, AST, alkaline phosphatase, LDH, glucose, sodium, potassium, chloride, CO2, calcium
○ Special tests; direct antiglobulin (DAT), serum free hemoglobin, haptoglobin
○ Urinalysis; including microscopic examination of urine sediment
○ Virology; retain samples for hepatitis A virus (HAV), HBV, HIV, B19V
- SC infusion and subject diary training (refer to section on “Subject diary”; includes but not limited to sites of administration, volume of infusion, concomitant medications, AE’s, number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment)
- Administer study product SCIG-x
- Record AE (subjects with SBI will be withdrawn from the study)
- Record number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
SC phase, infusion weeks 2 and 3
- Abbreviated physical examination (with inspection of prior injection sites), record vital signs
- Record concomitant medications
- SC infusion training
- Administer study product SCIG-x
- Review infusion details and diary
- Record AE (including SBI)
- Record number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
SC phase, infusion weeks 4, 6, 7, 8, 10, 11, 12, 15, 16, 18, 19, 20, 22, 23, 24 (home setting)
- Subject/caregiver administers study product SCIG-x
- Complete subject diary
SC phase, infusion weeks 5, 9, 14, 17, 21
- Abbreviated physical examination (with inspection of prior injection sites), body weight (for dose calculation, excluding visit 14), height, vital signs
- Record concomitant medications
- Take sample prior to infusion for trough total IgG
- Additional assessments prior to infusion (for infusions 9 and 17 only):
○ Hematology; including complete blood count, white blood differential and platelet count
○ Blood chemistry; including total bilirubin, creatinine, BUN, ALT, AST, alkaline phosphatase, LDH, glucose, sodium, potassium, chloride, CO2, calcium
○ Special tests; direct antiglobulin (DAT), serum free hemoglobin, haptoglobin
○ Urinalysis; including microscopic examination of urine sediment
○ IgG subclass and antibody titer levels for S. pneumoniae, H. influenzae, and C. tetani (within 0.5 hours before SCIG-x infusion)
- Subject/caregiver administers study product SCIG-x
- Review infusion details and diary
- Record AE (including SBI)
- Record number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
SC phase, infusion week 13 and PK assessment
- Abbreviated physical examination (with inspection of prior injection sites), body weight (for dose calculation), record vital signs
- Record concomitant medications
- Take sample prior to infusion for trough total IgG
- Subject/caregiver administers study product SCIG-x
- Review infusion details and diary
- Record AE (including SBI)
- Record number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
- Take samples for PK assessment of IgG levels at the following time points (exact time of sample collection to be recorded; only for subjects age >5 years):
○ Prior to infusion (within 0.5 hours; same as pre dose trough IgG sample)
○ Immediately following completion of infusion
○ Post infusion—1 day (±4 hours), 3 days (±4 hours), 4 days (±4 hours), 5 days (±4 hours), 7 days (±1 day; within 0.5 hours before infusion 14)
Completion of 24 weeks of SC infusions before PK analysis is complete
- Continue to receive weekly SC infusion of SCIG-x in the home setting
- Complete subject diary
- Return to clinic every 4 weeks:
○ Abbreviated physical examination (with inspection of prior injection sites), body weight (for dose calculations), height, vital signs
○ Record concomitant medications
○ Take sample prior to infusion for trough total IgG
○ Caregiver/subject to administer SCIG-x
○ Review infusion details and diary
○ Record AE (including SBI)
○ Record number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
- At 8 week intervals:
○ Hematology; including complete blood count, white blood differential and platelet count
○ Blood chemistry; including total bilirubin, creatinine, BUN, ALT, AST, alkaline phosphatase, LDH, glucose, sodium, potassium, chloride, CO2, calcium
○ DAT, serum free hemoglobin, haptoglobin
○ Urinalysis; including microscopic examination of urine sediment
○ IgG subclass and antibody titer levels for S. pneumoniae, H. influenzae, and C. tetani (within 0.5 hours before SCIG-x infusion)
Final study visit/early termination visit
- Full physical examination, record vital signs
- Record concomitant medications
- Blood and urine for clinical laboratory assessments:
○ Trough total IgG, IgG subclass, and antibody titer levels for measles, S. pneumoniae, H. influenzae, and C. tetani
○ Serum pregnancy test (females of child-bearing potential)
○ Hematology; including complete blood count, white blood differential and platelet count
○ Blood chemistry; including total bilirubin, creatinine, BUN, ALT, AST, alkaline phosphatase, LDH, glucose, sodium, potassium, chloride, CO2, calcium
○ Special tests; Direct antiglobulin (DAT), serum free hemoglobin, haptoglobin
○ Urinalysis; including microscopic examination of urine sediment
- Record AE (subjects with SBI will be withdrawn from the study)
- Record number of days of missed work/school/kindergarten/daycare/daily activities due to infections and related treatment
- Review subject diary
Assessment of pharmacokinetics
Primary PK objective
Total IgG concentrations will be tabulated for IV and SC modes of administration, with individual as well as mean total IgG concentrations vs. time curves established. AUC, Cmax, and Tmax will be determined. To meet the primary PK endpoint, non-inferiority of the steady-state IgG AUC between SC dose (AUC0-t,SC; established from SC infusion 13) of SCIG-x and IV dose (AUC0-t,IV; established from IV infusion 1) of IVIG-x will be tested using established protocols for bioequivalence, and the 90% confidence interval of the geometric LSM ratio of SC AUC and IV AUC compared.
Secondary PK objective
Steady-state mean trough concentrations of total IgG will be determined from the average of Ctrough measurements at SC infusions 13, 14, 17, and 21 (for SC administration), and IV infusion 1 and immediately prior to IV infusion 2 (for IV administration).
Exploratory PK objectives
Trough levels of IgG subclasses IgG1, IgG2, IgG3, IgG4, as well as antibody titres against S. pneumoniae, H. influenzae, and C. tetani will be determined for IV and SC methods of administration.
Assessment of efficacy
Exploratory variables assessing the efficacy of SCIG-x study drug include:
- Rate of SBIs: bacteremia/sepsis, bacterial meningitis, osteomyelitis/septic arthritis, bacterial pneumonia, visceral abscess
- Rate of infections: serious and non-serious (including acute sinusitis, worsening of chronic sinusitis, otitis media, pneumonia, bronchitis and pathogen-associated diarrhea), as well as infections validated by positive radiograph, fever, culture, or diagnostic testing
- Days of missed work/school/kindergarten/daycare/daily activities due to infections and associated treatments
- Days on antibiotics (prophylactic as well as for treatment of infection)
- Number of hospitalizations due to infections
Assessment of safety
Safety analysis will include summaries of:
- Adverse events (encompassing adverse reactions, suspected adverse reactions, unexpected AE, serious AE), including determination of severity (mild, moderate, severe) and causality (certain, probably, possible, not related), to be presented as the number of adverse events and number and percentage of subjects with adverse events
- Subjects who discontinued due to death, adverse events or serious adverse events
- Adverse events taking place within 72 hours of completion of study drug administration
- Local infusion site reactions
Safety parameters
Results of all laboratory tests and vital signs will be summarized:
- Vital signs (SBP, DBP, HR, T, RR)
- Routine blood samples
○ Hematology: hemoglobin, hematocrit, platelets, red blood cell count and morphology, white blood cell count, and differential counts
○ Blood chemistry: total bilirubin, indirect bilirubin, creatinine, BUN, ALT, ALP, AST, alkaline phosphatase, LDH, glucose, sodium, potassium, chloride, CO2, calcium, bicarbonate
- Urinalysis: pH, protein, glucose, ketones, bilirubin, nitrites, urobilinogen, blood, leukocyte esterase (including microscropic examination of urine sediment if abnormal)
- Virology: HAV, HBV, HCV, HIV, B19V (these virus safety retain samples will be stored for possible future testing)
- Special tests: DAT, serum-free hemoglobin, haptoglobin
REFERENCES
Berger M. and Allen J.A. 2015. Optimizing IgG therapy in chronic autoimmune neuropathies: A hypothesis driven approach. Muscle Nerve. 51:315–326.
Berger M., Rojavin M., Kiessling P., and Zenker O. 2011. Pharmacokinetics of subcutaneous immunoglobulin and their use in dosing of replacement therapy in patients with primary immunodeficiencies. Clin. Immunol. 139:133–141.
Berger M., Jolles S., Orange J.S., and Sleasman J.W. 2013. Bioavailability of IgG administered by the subcutaneous route. J. Clin. Immunol. 33:984–990.
Bonilla F.A. and Geha R.S. 2003. 12. Primary immunodeficiency diseases. J. Allergy Clin. Immunol. 111:S571–S581.
Bruton O.C. 1952. Agammaglobulinemia. Pediatrics. 9:722–728.
Ochs H.D., Gupta S., Kiessling P., Nicolay U., Berger M., and the Subcutaneous IgG Study Group. 2006. Safety and efficacy of self-administered subcutaneous immunoglobulin in patients with primary immunodeficiency diseases. J. Clin. Immunol. 26:265–273.
Orange J.S., Belohradsky B.H., Berger M., Borte M., Hagan J., Jolles S., Wasserman R.L., Baggish J.S., Saunders R., and Grimbacher B. 2012. Evaluation of correlation between dose and clinical outcomes in subcutaneous immunoglobulin replacement therapy. Clin. Exp. Immunol. 169:172–181.
Picard C., Al-Herz W., Bousfiha A., Casanova J.L., Chatila T., Conley M.E., Cunningham-Rundles C., Etzioni A., Holland S.M., Klein C., Nonoyama S., Ochs H.D., Oksenhendler E., Puck J.M., Sullivan K.E., Tang M.L., Franco J.L., and Gaspar H.B. 2015. Primary immunodeficiency diseases: An update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J. Clin. Immunol. 35:696–726.
Radinsky S. and Bonagura V.R. 2003. Subcutaneous immunoglobulin infusion as an alternative to intravenous immunoglobulin. J. Allergy Clin. Immunol. 112:630–633.
Resnick E.S. and Cunningham-Rundles C. 2012. The many faces of the clinical picture of common variable immune deficiency. Curr. Opin. Allergy Clin. Immunol. 12:595–601.
Roifman C.M. and Gelfand E.W. 1988. Replacement therapy with high dose intravenous gamma-globulin improves chronic sinopulmonary disease in patients with hypogammaglobulinemia. Pediatr. Infect. Dis. J. 7:S92–S96.
Stiehm E.R., Casillas A.M., Finkelstein J.Z., Gallagher K.T., Groncy P.M., Kobayashi R.H., Oleske J.M., Roberts R.L., Sandberg E.T., and Wakim M.E. 1998. Slow subcutaneous human intravenous immunoglobulin in the treatment of antibody immunodeficiency: Use of an old method with a new product. J. Allergy Clin. Immunol. 101:848–849.
Wang W., Wang E.Q., and Balthasar J.P. 2008. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 84:548–558.
Wasserman R.L., Irani A.M., Tracy J., Tsoukas C., Stark D., Levy R., Chen J., Sorrells S., Roberts R., and Gupta S. 2010. Pharmacokinetics and safety of subcutaneous immune globulin (human), 10% caprylate/chromatography purified in patients with primary immunodeficiency disease. Clin. Exp. Immunol. 161:518–526.
Wasserman R.L., Melamed I., Nelson R.P. Jr., Knutsen A.P., Fasano M.B., Stein M.R., Rojavin M.A., and Church J.A. 2011. Pharmacokinetics of subcutaneous IgPro20 in patients with primary immunodeficiency. Clin. Pharmacokinet. 50:405–414.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 6 • Number 2 • June 2019
Pages: 75 - 86
History
Received: 11 April 2019
Accepted: 16 May 2019
Accepted manuscript online: 22 May 2019
Copyright
© 2019.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
LindaVong. 2019. A clinical trial protocol to evaluate the safety and pharmacokinetics of subcutaneously administered immunoglobulin in patients with primary immunodeficiency. LymphoSign Journal.
6(2): 75-86. https://doi.org/10.14785/lymphosign-2019-0005
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
