Leukocyte adhesion deficiency-I caused by a novel mutation in ITGB2 presenting with pyoderma gangrenosum
Abstract
Background: Leukocyte adhesion deficiency (LAD) syndromes are primary immunodeficiency disorders caused by defects in adhesion molecules on leukocytes resulting in impaired migration into tissues. Common cutaneous manifestations of LAD include bacterial infections, omphalitis with delayed separation of the umbilical cord, impaired pus formation and poor wound healing. LAD is associated with significant morbidity and mortality, making early diagnosis and management integral in the care of these patients.
Methods: Molecular testing and flow cytometry for expression of CD18 were performed on 2 siblings presenting with cutaneous lesions including pyoderma gangrenosum (PG).
Results: We describe 2 siblings with a novel homozygous mutation in ITGB2 (c.2070del, p.Asp690Glufs*25) resulting in an atypical presentation of LAD-I with PG.
Conclusion: LAD should be considered in patients presenting with unexplained PG, even in the absence of significant infections or umbilical cord complications.
Statement of novelty: To the best of our knowledge, we describe a novel homozygous mutation in ITGB2 (c.2070del, p.Asp690Glufs*25) resulting in LAD-I. Patients with LAD-I may present with unexplained PG and may lack classic symptoms including umbilical cord complications.
Introduction
The leukocyte adhesion cascade system allows for leukocyte accumulation at sites of tissue inflammation and infection. Adhesion molecules including selectins, integrins, and members of the immunoglobulin superfamily of proteins, are expressed on leukocytes and vascular endothelial cells and are required for leukocytes to migrate from the vasculature into the tissues (Schmidt et al. 2013). Leukocyte adhesion deficiency (LAD) syndromes are primary immunodeficiency disorders caused by defects in adhesion molecules on leukocytes resulting in impaired migration into tissues (Al-Herz et al. 2011). LAD is divided into 3 subgroups including LAD-I (beta-2 integrin defect), LAD-II (fucosylated carbohydrate ligands for selectins are absent) and LAD-III (activation of all beta integrins is defective) (Al-Herz et al. 2011). LAD-I is an autosomal recessive syndrome caused by a mutation in the integrin beta-2 gene (ITGB2) resulting in deficiency and/or defects in CD18, the common beta chain of the beta-2 integrin family, and the inability of leukocytes to adhere to the endothelium and migrate into tissues (van de Vijver et al. 2012). LAD-I has also been associated with impaired inhibition of interleukin-23 and interleukin-17 resulting in a hyperinflammatory response and chronic inflammation (Moutsopoulos et al. 2014). Patients with severe LAD-I, defined as less than 2% of CD18 expression, typically present in early infancy with recurrent, life threatening infections that are frequently fatal before age 2 years old without hematopoietic stem cell transplant (HSCT) (Almarza Novoa et al. 2018). Patients with mild to moderate LAD-I, defined as 2%–30% of CD18 expression, tend to have fewer significant infections and often survive into adulthood without HSCT (Almarza Novoa et al. 2018). Common clinical manifestations of LAD-I include delayed separation of the umbilical cord with omphalitis, recurrent bacterial infections especially of skin and mucus membranes, impaired wound healing, absent pus formation, periodontitis, and leukocytosis (Hanna and Etzioni 2012). We describe 2 siblings with LAD-I caused by a novel mutation in ITGB2 and atypical presentation including pyoderma gangrenosum (PG).
Methods
Molecular genetic evaluation
DNA collected from blood was submitted to Fulgent Diagnostics for sequencing. DNA was barcoded and enriched for the coding exons of targeted genes using hybrid capture technology. Prepared DNA libraries were sequenced using Next Generation Sequencing technology. Following alignment, variants were detected and interpreted manually using locus specific databases, literature searches and other molecular biological principles. All variants were confirmed by Sanger sequencing. Patient consent was obtained and recorded through the Canadian Primary Immunodeficiency Evaluation Study (C-PRIMES).
Flow cytometry
Flow cytometric immunophenotyping of peripheral blood was performed by Mayo Medical Laboratories to evaluate the presence of the CD11/CD18 complex using monoclonal antibodies directed against the CD11 isoforms, CD11a and CD11b, and CD18 antigens.
Results
Patient presentation
A female patient born to non-consanguineous Caucasian parents presented at age 7 years old with multiple painful, erythematous and necrotic papules on the inner thigh and intermittent fevers. She was admitted for intravenous antibiotics following a failed course of oral cephalexin. Wound and blood cultures were negative. She had a significant lymphocytosis, 35.1 × 109/L, with predominate neutrophils, 28.4 × 109/L. Past infectious history included 1 previous cutaneous infection following an abrasion and molluscum contagiosum. She did not have delayed separation of the umbilical cord. Partial evaluation of the immune system was performed at age 7 years old and reported normal lymphocyte subsets (CD20+, CD3+, CD3+/CD4+, CD3+/CD8+, and CD3−/CD56+CD16+), normal immunoglobulin levels, normal vaccine titers, and normal neutrophil oxidative burst index. The patient had a persistent leukocytosis ranging from 38.8–10.8 × 109/L and neutrophilia ranging from 32.6–11.3 × 109/L.
Fifteen months later, she presented to the emergency room with papules that rapidly progressed to necrotic ulcerations on the thighs and fever requiring another admission for intravenous antibiotics (Figures 1–4). Dermatology was consulted and a skin biopsy was performed. The skin biopsy reported epidermal necrosis with superficial dermal abscess with neutrophils and eosinophils consistent with superficial granulomatous pyoderma. Evaluations for inflammatory bowel disease, including colonoscopy with biopsies, and rheumatologic disorders were negative and the patient was treated for presumed idiopathic PG with oral prednisone and cyclosporine. Genetics was consulted due to the persistent nature and difficult to treat skin lesions. A literature review led to the article by Madkaikar et al, describing patients with PG in the setting of persistent neutrophilia who were diagnosed with LAD-1. Given this, at age 9 years and 6 months, molecular genetic testing was arranged and a homozygous frameshift mutation was identified in the ITGB2 gene (c.2070del, p. Asp690Glufs*25) located on 21q22.3. This was predicted to result in premature truncation of the protein and according to the American College of Medical Genetics (ACMG) guidelines the variant was classified as pathogenic. A heterozygous variant in NOD2 (c.3019dup, p.Leu1007Profs*2) was also identified and was classified as a susceptibility factor for Crohn’s disease. Flow cytometry reported absent expression of CD18 on lymphocytes with significantly reduced expression on granulocytes. There was normal expression of CD11a on lymphocytes and monocytes and absent expression on granulocytes. CD11b was absent on lymphocytes, significantly reduced in monocytes, and close to normal on granulocytes. The patient was diagnosed with LAD-I. Immunosuppressive medications were discontinued and she was started on prophylactic antibiotics. The PG has resolved since starting prophylactic antibiotics with amoxicillin. She has had intermittent skin lesions that resolve with a course of oral antibiotics.
Figure 1:
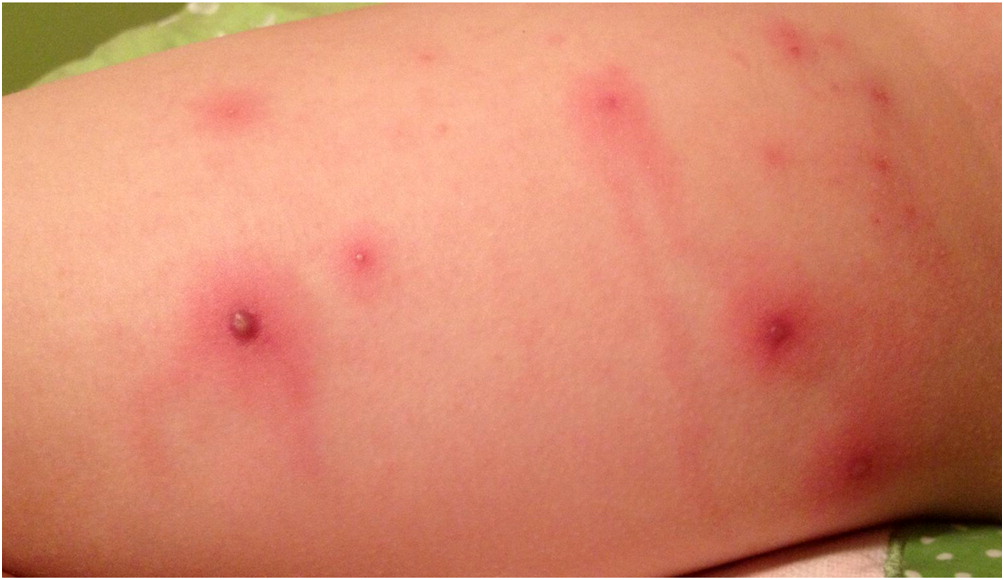
Figure 2:
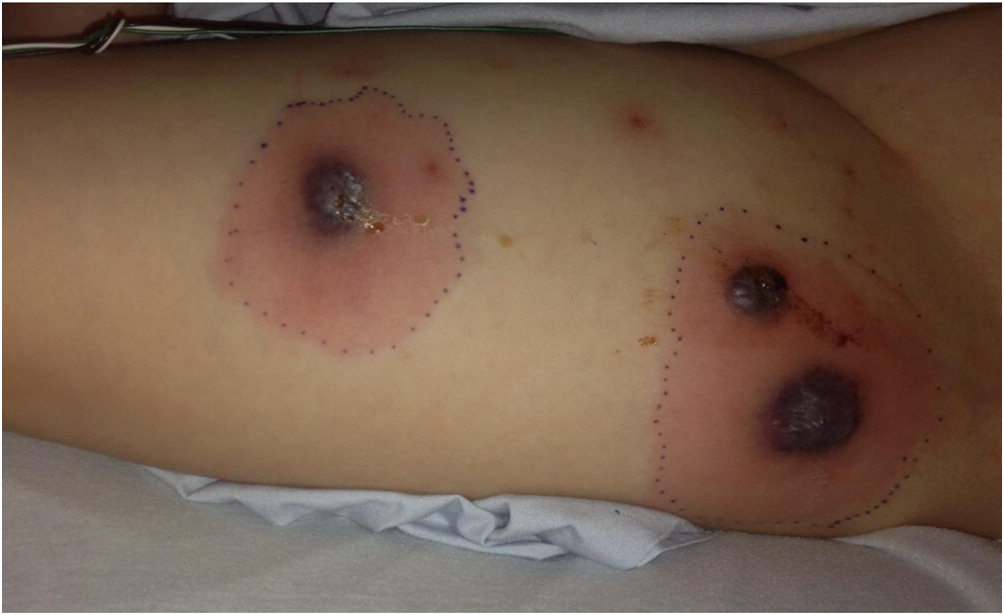
Figure 3:

Figure 4:

Patient 2, the brother of patient 1, presented at age 13 years with small painful papules on the lateral aspect of upper thigh. Past infectious history was remarkable for molluscum contagiosum. He did not have delayed separation of the umbilical cord. Based on the sibling’s diagnosis of LAD-I, molecular genetic testing was completed and he was confirmed to have the same homozygous mutation in ITGB2 (c.2070del, p.Asp690Glufs*25). The parents of patient 1 and patient 2 also had molecular genetics testing completed and were both found to be carriers of the mutation in ITGB2. The patient was diagnosed with LAD-I and was started on prophylactic antibiotics to prevent the development of cutaneous infections. The papules have resolved since starting prophylactic antibiotics.
Discussion
LAD-I is a rare primary immunodeficiency caused by defects in adhesion molecules on leukocytes resulting in impaired migration into tissues. To the best of our knowledge, we describe a novel homozygous variant in ITGB2 (c.2070del, p.Asp690Glufs*25) resulting in LAD-I. Common cutaneous and mucus membrane manifestations of LAD-I include bacterial infections, omphalitis with delayed separation of the umbilical cord, gingivitis, oral ulcers, impaired pus formation, and poor wound healing. In particular, necrotic skin lesions have been reported in more than 10% of patients with LAD-I (Almarza Novoa et al. 2018). PG is an ulcerative skin disease characterized by sterile, painful, necrotic ulcers and is commonly associated with systemic conditions including inflammatory bowel disease, hematologic disorders, and rheumatic disorders (Wollina 2007). Patients with LAD-I presenting predominately with PG have been reported (Van de Kerkhof and Weemaes 1990; Bedlow et al. 1998; Hinze et al. 2010; Nord et al. 2011; Thakur et al. 2013; Madkaikar et al. 2015). Similar to patient 1, most of these reported patients did not have significant infections prior to presenting with PG and therefore the diagnosis of LAD-I was often delayed. The histopathology of classic PG shows predominant neutrophil infiltration. In contrast, the histopathology of LAD-I ulcerating lesions typically has an absence of neutrophils (Bedlow et al. 1998; Nord et al. 2011). The skin biopsy of patient 1 reported the presence of neutrophils suggesting partial neutrophil recruitment into the tissues. Similar histopathology findings have been described in patients with LAD-I and PG (Hinze et al. 2010; Madkaikar et al. 2015). This case report highlights the importance of considering LAD-I in patients presenting with PG, even in the absence of significant infections.
Other atypical features of our LAD-I patients include lack of umbilical cord complications and older age of presentation. Umbilical cord complications such as delayed separation of the umbilical cord and/or omphalitis are early manifestations of LAD-I. Umbilical cord complications are more common in patients with severe LAD-I (84% of patients) compared to mild to moderate LAD-I (58% of patients) (Almarza Novoa et al. 2018). Neither of our patients had umbilical cord complications. In addition, our patients presented later in childhood, at ages 7 and 13 years old, compared to the majority of other LAD-I patients described in the literature. Almarza Novoa et al. 2018 reported the median age of presentation was 1 month old (range 0.03–18 months) for severe LAD-I and 6 months old (range 0.03–192 months) for mild to moderate LAD-1. The lack of umbilical cord complications and older age at the time of presentation may have contributed to the delayed diagnosis of LAD-I in patient 1. In addition, it could be hypothesized that this particular mutation results in a mild phenotype explaining the delayed onset of presentation.
We present 2 siblings with a novel homozygous mutation in ITGB2 (c.2070del, p.Asp690Glufs*25) resulting in LAD-I with atypical presentation including PG. LAD-I should be considered in patients with unexplained PG, even in the absence of classic symptoms such as umbilical cord complications. An early diagnosis of LAD-I is imperative to prevent life-threatening complications such as significant infections and for consideration of treatments including HSCT.
REFERENCES
Al-Herz W., Bousfiha A., Casanova J.L., Chapel H., Conley M.E., Cunningham-Rundles C., Etzioni A., Fischer A., Franco J.L., Geha R.S., Hammarstrom L., Nonoyama S., Notarangelo L.D., Ochs H.D., Puck J.M., Roifman C.M., Seger R., and Tang M.L.K. 2011. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies expert committee for primary immunodeficiency. Front Immunol. 2:1–26.
Almarza Novoa, E., Kasbekar, S., Thrasher, A.J., Kohn, D.B., Sevilla, J., Nguyen, T., Schwartz, J.D., and Bueren, J.A. 2018. Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J. Allergy Clin. Immunol. Pract. (In press).
Bedlow A.J., Davies E.G., Moss A.L.H., Rebuck N., Finn A., and Marsden R.A. 1998. Pyoderma gangrenosum in a child with congenital partial deficiency of leukocyte adhesion. British J. Dermatol. 139(6):1064–1067.
Hanna S. and Etzioni A. 2012. Leukocyte adhesion deficiencies. Ann. N. Y. Acad. Sci. 1250:50–55.
Hinze C.H., Lucky A.W., Bove K.E., Marsh R.A., Bleesing J.H., and Passo M.H. 2010. Leukocyte adhesion deficiency type 1 presenting with recurrent pyoderma gangrenosum and flaccid scarring. Pediatr. Dermatol. 27: 500–503.
Madkaikar M., Italia K., Gupta M., Desai M., Aggarwal A., Singh S., Suri D., Mishra A., Chavan S., Ghosh K., Sarangal R., and Dogra S. 2015. Leukocyte adhesion deficiency with a novel intronic mutation presenting with pyoderma gangrenosum. J. Clin. Immunl. 35:431–434.
Moutsopoulos N.M., Konkel J., Sarmadi M., Eskan M.A., Wild T., Dutzan N., Abusleme L., Zenobia C., Hosur K.B., Abe T., Uzel G., Chen W., Chavakis T., Holland S.M., and Hajishengallis G. 2014. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci. Transl. Med. 6(229):229ra40.
Nord K.M., Pappert A.S., and Grossman M.E. 2011. Pyoderma gangrenosum-like lesions in leukocyte adhesion deficiency I treated with intravenous immunoglobulin. Pediatr. Dermatol. 28(2):156–161.
Schmidt S., Moser M., and Sperandio M. 2013. The molecular basis of leukocyte recruitment and its deficiencies. Mol. Immunol. 55:49–58.
Thakur H., Sodani R., Chandra J., and Singh V. 2013. Leukocyte adhesion defect type 1 presenting with recurrent pyoderma gangrenosum. Indian J Dermatol. 58:158.
Van de Kerkhof P.C.M. and Weemaes C.M.R. 1990. Skin manifestations in congenital deficiencies of leukocyte-adherence glycoproteins. British J Dermatol. 123:395–401.
Van de Vijver E., Maddalena A., Sanal O., Holland S.M., Uzel G., Madkaikar M., de Boer M., van Leeuwen K., Köker M.Y., Parvaneh N., Fischer A., Law S.K., Klein N., Tezcan F.I., Unal E., Patiroglu T., Belohradsky B.H., Schwartz K., Somech R., Kuijpers T.W., and Roos D. 2012. Hematologically important mutations: leukocyte adhesion deficiency (first update). Blood Cells Mol. Dis. 48:53–61.
Wollina U. 2007. Pyoderma gangrenosum—A review. Orphanet J. Rare Dis. 2:19.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 5 • Number 3 • September 2018
Pages: 86 - 90
History
Received: 15 May 2018
Accepted: 5 July 2018
Accepted manuscript online: 30 July 2018
Copyright
© 2018.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AlisonHaynes, AndrewO’Keefe, PaulDancey, KamalOhson, LesleyTurner, and MarisaChard. 2018. Leukocyte adhesion deficiency-I caused by a novel mutation in ITGB2 presenting with pyoderma gangrenosum. LymphoSign Journal.
5(3): 86-90. https://doi.org/10.14785/lymphosign-2018-0006
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
