Advances in the diagnosis and management of haemophagocytic lymphohistiocytosis: a review of literature
Abstract
Haemophagocytic lymphohistiocytosis (HLH) is a life-threatening syndrome, triggered by the excessive stimulation of lymphocytes and macrophages producing abnormally increased levels of cytokines. The diagnosis can be challenging due to overlapping signs and symptoms with other diseases. Therefore, early detection and prompt initiation of treatment is crucial for better survival. There are mainly 2 forms of HLH, primary (genetic) and secondary (acquired). Recent advances in the diagnosis and treatment modalities have led to better understanding of HLH. Here, we present a concise review of the literature with recent updates in pathogenesis, diagnostic modalities, and treatment of HLH.
Statement of novelty: In this concise review of HLH, we discuss from the historical background to contemporary diagnostic modalities, as well as recent updates on treatment.
Background
Haemophagocytic lymphohistiocytosis (HLH) is a life-threatening disorder caused by a hyper-inflammatory response. It is triggered by excessive activation of lymphocytes and macrophages producing elevated levels of cytokines (Janka 2007b). HLH is classified into 2 major forms which are difficult to distinguish clinically: primary (genetic) and secondary (acquired). Primary HLH typically manifests in children with underlying genetic abnormalities. It is further sub-classified into two types, familial haemophagocytic lymphohistiocytosis (FHLH), and the other associated with underlying primary immunodeficiency syndromes. Secondary HLH is categorized according to the underlying etiology, such as infections or malignancy associated HLH (Gupta and Weitzman 2010).
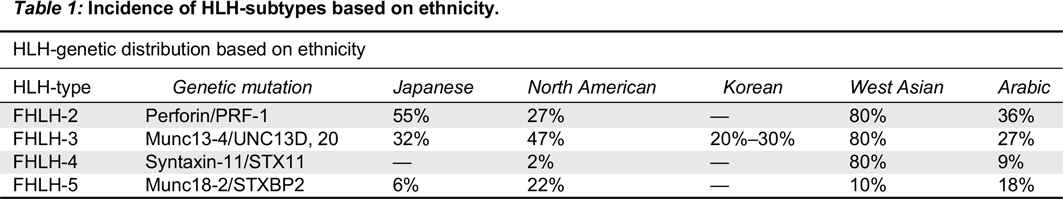
HLH can affect all age groups with no race or sex predilection (Janka 2012). As HLH is potentially fatal in the absence of appropriate treatment, a high index of suspicion is required for early diagnosis and treatment. The exact incidence of HLH is not known, however, estimates of 0.12–0.34/million children under 15 years of age have been noted in the literature (Henter et al. 1991a; Ishii et al. 1998). The incidence of different genetic types of HLH varies among countries (Nagai et al. 2010) (Table 1). A retrospective analysis from Texas estimated the prevalence to be nearly 1 in 100 000 children younger than 18 years of age (Niece et al. 2010). A much higher occurrence of 7.5 in 10 000 hospitalized patients was reported from Turkey, and may be associated with the extraordinary prevalence of consanguinity in the Turkish community (Gürgey et al. 2003).
Table 1:
Historical background
In 1939, Scott and Robb-Smith described a sporadic disease presenting with fever, lymphadenopathy, and massive hepatosplenomegaly, and named it histiocytic medullary reticulosis (Scott and Robb-Smith 1939). Later, Farquhar and Claireux in 1952 recognized the constellation of signs and symptoms in four siblings, of whom 3 succumbed to their disease. They reported the condition as familial haemophagocytic reticulosis based on widespread proliferation of histiocytes throughout the reticuloendothelial system and haemophagocytosis (Farquhar and Claireaux 1952; Farquhar et al. 1958). Since then, much progress has been made in identifying the pathophysiology and genetic mutations responsible for the disease.
Pathophysiology
The histological hallmark of HLH is an accumulation of histiocytes in various tissues along with defective cytotoxic function. In immunocompetent individuals, cytotoxic T-lymphocytes (CTL), natural killer (NK) cells, and T-regulatory cells are critical for the control of infectious and inflammatory responses. The innate immune system acts as the first line of defense against infections. It is mediated by phagocytes, neutrophils, and NK cells which together initiate phagocytosis, antigen presentation, and activation of the adaptive immune system. The pathogen infected “target cells” are recognized by NK cells and CTLs. The CTLs, with helper T cell receptors (TCRs), initiate the cascade of immune response. This requires effective cell lysis through granule-mediated cytotoxicity. The granules are transported to the cell surface by the process of exocytosis, and are released into the target cells. Entry of the granules from activated CTLs and NK cells into the target cell is facilitated by perforin, which is produced by NK and CD8+ T cells. Once perforin is liberated, pores are formed in the target cell membrane, allowing granzyme-B to pass into the target cell and induce cell apoptosis/death (Figure 1). HLH arises from the impaired continuous stimulation of CD8+ T cells, which in turn produces excessive INF-γ and further promotes uncontrolled secretion of other cytokines such as IL-1, IL-6, IL-10, IL-18, and TNF-α (Janka and Lehmberg 2014). The defective function of NK cells and CTLs is permanent in primary HLH and transient in secondary HLH (Henter et al. 1991c; Faitelson and Grunebaum 2014; Brisse et al. 2015).
Figure 1:
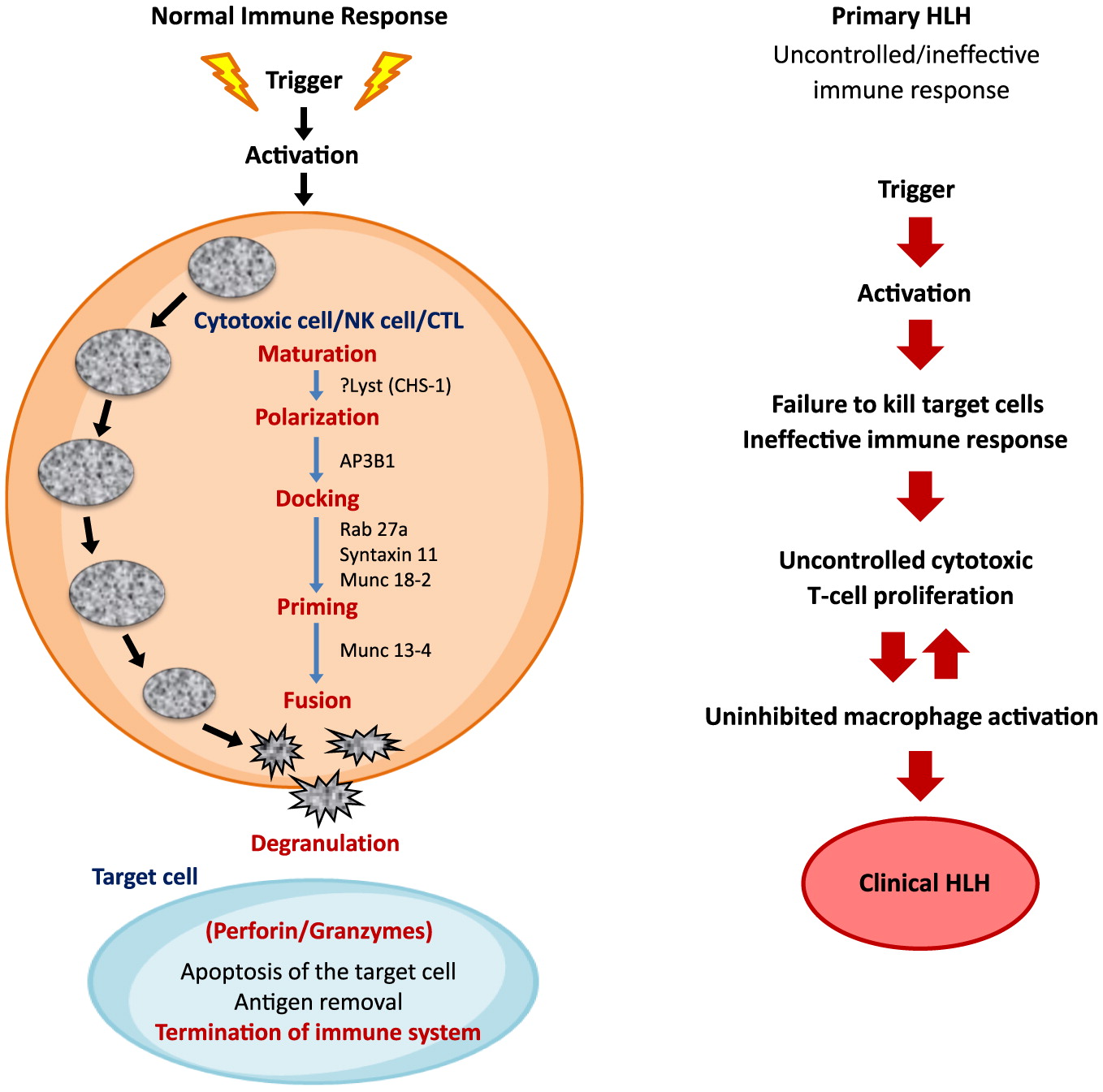
The circulating high concentrations of inflammatory cytokines and infiltration of the organs by activated lymphocytes and histiocytes manifest clinically as prolonged fever, hepatosplenomegaly, cytopenias, and (or) multi-organ failure (George 2014; Madkaikar et al. 2016). Fever is induced by IL-1, IL-6, and TNF-α. Similarly, pancytopenia is a result of suppressive activity of TNF-α and INF-γ, along with hemophagocytosis and elevated transaminases, bilirubin, and lactate dehydrogenase (LDH). Low concentration of fibrinogen in blood is a result of high circulating levels of plasminogen activator secreted by the activated macrophages. This further stimulates plasmin, consequently causing hyperfibrinolysis. High concentration of triglyceride is a result of the suppressive actions of TNF-α on lipoprotein lipase. Similarly, activated histiocytes/macrophages lead to hyperfibrinolysis resulting in high serum ferritin (Janka and Lehmberg 2014; Yang et al. 2016). Growth differentiation factor (GDF15)-mediated upregulation of ferroportins may also contribute in hyperferritinemia (Wu et al. 2013).
Primary HLH
Clinically, primary HLH is indistinguishable from the secondary form. In some cases, the diagnosis of FHLH is presumed by presentation at an early age, unremitting course or multiple reactivations, and presence of a similar history in one or more family members (Arico et al. 1996).
Genetic HLH
Genetic HLH is linked to distinct gene abnormalities in different ethnicities (Ohadi et al. 1999; Gürgey et al. 2003; Côte et al. 2009; Danielian et al. 2010; Marsh et al. 2010a, 2010b; Nagai et al. 2010; Yoon et al. 2010; Rosado and Kim 2013) (Table 2). The molecular basis of FHLH is heterogeneous, with defects occurring in one or more of several proteins in the NK/CTL pathway; from maturation, polarization, docking, and priming, to fusion and degranulation (Usmani et al. 2013). The protein products of these genes are responsible for the commencement to completion of CTLs and NK cell cytolytic activity. Any mutation of these genes that affect the pathway of the respective protein products leads to significant hypercytokinemia and defective immune response.
Table 2:

Note: There are many primary immunodeficiency syndromes associated with HLH.
FHL-1, linked to chromosome 9q21.3-22, was first described in 4 Pakistani and a Saudi family. However, the corresponding gene and associated protein remained unidentified (Ohadi et al. 1999). In 1999, Stepp et al. described the association of perforin gene mutation to HLH (FHLH-2) (Stepp et al. 1999). Subsequently, 3 more genetic defects have been identified which play a key role in the cytotoxic granule pathway (FHL-3, 4, and 5) (Feldmann et al. 2003; zur Stadt et al. 2005; Côte et al. 2009; Macartney et al. 2011). All the above mutations are transmitted in an autosomal recessive manner. A report from Zhang et al. describes synergistic defects of different disease-causing mutations leading to FHLH, suggesting a potential digenic mode of inheritance (Zhang et al. 2014).
Other inherited immune deficiency syndromes associated with life-threatening HLH include pigmentary and non-pigmentary disorders. The pigmentary immune deficiency disorders include Griscelli syndrome-type 2 (GS-2), Chediak-Higashi syndrome-1(CHS-1), and Hermansky-Pudlak syndrome-2 (HPS-2) are linked to RAB27A, LYST, and AP3B1 genetic mutations, respectively. CHS-1 presents with oculocutaneous albinism, easy bruising, and associated frequent bacterial infections. The clinical picture of HLH in CHS-1 is described as “accelerated phase”, mimicking acquired HLH (Janka 2007a). HPS-2 is characterized by oculocutaneous albinism, bleeding, and neutropenia (Gholam et al. 2011).
The non-pigmentary immune deficiency syndromes associated with HLH are mainly X-linked lymphoproliferative (XLP) disorders. The genetic mutation of XLP-1 is linked to the Src-homology 2 domain-containing gene (SH2D1A), a lymphocyte signaling adapter. XLP-2 is caused by a gene mutation in BIRC4, which encodes X-linked inhibitor of apoptosis (XIAP). Both XLP-1 and XLP-2 syndromes have a high predilection to Epstein-Barr virus (EBV) infection, HLH, and dysgammaglobulinemia (Yang et al. 2012). There is an association of lymphomas with XLP-1 but not XLP-2. Patients with XLP-2 develop colitis and can be at risk of being misdiagnosed with Crohn’s disease. A novel mutation in IL-2-ITK gene has been observed in few patients, both boys and girls with EBV-lymphoproliferative disease and HLH (Ghosh et al. 2014). A recent review of 8 pediatric intensive care unit patients diagnosed with EBV-HLH found 50% to be associated with an ITK mutation, representing a very poor prognosis (Zheng et al. 2016).
Primary immunodeficiency disorders (PID) and HLH
Although infrequent, HLH has been described in patients with different types of PID, particularly those related to genetic T cell, NK cell, and few B-cell/macrophage deficiencies. It is prudent to remember that acquired HLH patient might have underlying PID. In an international survey from centers involved in the diagnosis and treatment of primary immune deficiencies and/or HLH (Histiocyte Society, European Society of Blood and Bone Marrow Transplantation’s inborn errors Working Party, and the German Society for Pediatric Oncology and Hematology), 63 patients with PID were identified who fulfilled the diagnostic criteria for HLH. More than 80% of patients had 2 main groups of PID; combined immune deficiencies (CID) and chronic granulomatous disease (CGD). Some of the patients in the CID group developed HLH in the first months of life, at or before the diagnosis of immune deficiency. Most of the patients with CGD presented beyond the first year of life, some in early adulthood (Bode et al. 2015).
In T cell defects, HLH is not associated with macrophage dysfunctions but primarily to T cell functional abnormality. HLH is seen in patients with classical severe combined immunodeficiency (SCID) lacking complete T cell function, which predisposes these patients to more infections and subsequent HLH. Regardless of the mechanism of immune dysfunction pertaining to B or T cells, clinicians should have a high index of suspicion to investigate for HLH or macrophage activation syndrome (MAS) (Faitelson and Grunebaum 2014).
Secondary HLH
Secondary HLH is seen in some patients with infections, malignancy, and those on immunosuppressive agents post-hematopoietic stem cell transplant (HSCT). The other known condition similar to HLH but distinct is macrophage activation syndrome (MAS) associated with autoimmune diseases, such as systemic lupus erythematosus (SLE), systemic juvenile idiopathic arthritis (sJIA) and adult-onset Still disease (Ramos-Casals et al. 2014; Degar 2015).
Infections associated haemophagocytic syndrome (IAHS)
It is now well established that many infectious agents including viruses, bacteria, protozoa, and fungi can trigger HLH (Janka et al. 1998; Rouphael et al. 2007). The first description of virus-associated haemophagocytic syndrome was given in 1979 by Risdall and colleagues on 19 immunocompromised patients, who developed haemophagocytic syndrome (Risdall et al. 1979). For unknown reasons IAHS is more commonly seen in adults than children (Falini et al. 1990). EBV may cause severe immune dysfunction leading to overwhelming infectious mononucleosis, lymphoproliferative disorders, dysgammaglobulinemia and HLH (Parvaneh et al. 2013). EBV-HLH has a high prevalence in Asian countries. A nation-wide survey from Japan in 2007 revealed more than 40% of patients diagnosed with EBV-HLH (Ishii et al. 2007). A review of 219 Japanese children from 1979–1995 showed IAHS related overall mortality of 52%. The main organism identified for the mortality was EBV, in approximately 73% of cases (72 of 99 children with EBV) (Janka et al. 1998).
Malignancy-associated HLH
HLH has been reported in association with various malignancies, primarily with T and/or NK cell lymphomas/leukemias, mainly in adults. However, it has also been reported in children with precursor-B acute lymphoblastic leukemia, myeloid leukemia, germ cell tumors and rarely with other solid malignancies (Moritake et al. 2014; Murphy et al. 2015). The most common triggering factor for HLH in these patients is an associated infection (viral, bacterial, fungal, or protozoal) in an already dysfunctional immune system possibly due to chemotherapy and/or malignancy (Jordan et al. 2011). Acquired HLH is also reported in patients receiving immunosuppressive therapy post-HSCT (Janka 2012). A nationwide retrospective study conducted by the Japanese Society of Pediatric Hematology (JPSH) reported 37 out of 42 patients from 1998–2008 fulfilled the inclusion criteria for post-HSCT-HLH. 26 patients had early onset HLH (defined as onset <30 days’ post-HSCT) with complete resolution in 69% (18/26) of patients. Patients with non-resolving HLH had a worse outcome (14%) versus those where the disease responded to treatment (59%) (Asano et al. 2012).
Macrophage activation syndrome (MAS)
MAS is a condition similar to HLH associated with autoimmune disorders, such as systemic juvenile idiopathic arthritis (sJIA), systemic lupus erythematosus (SLE) or other rheumatic diseases. MAS can lead to significant morbidity and mortality in pediatric rheumatology patients. The clinical manifestations are similar to HLH, with severe and potentially fatal hypercytokinemia but the mortality is higher in FHLH (Grom 2017).
Most MAS cases have been described mainly in sJIA in its adult-onset form, but also in SLE and occasionally in Kawasaki disease (Kim et al. 2011). MAS can be a presenting feature of sJIA, rendering the distinction from classical HLH difficult, especially in the absence of arthritis as a presenting symptom (Grom and Mellins 2010). A study on 14 patients with sJIA and MAS by Kaufman et al. used whole exome sequencing and identified rare protein altering variants in known HLH-associated genes, supporting the cytolytic pathway in MAS and in addition genetic overlap between MAS in sJIA and FHL (Kaufman et al. 2014).
Acute liver failure (ALF) and secondary HLH
Some cases of acute liver failure are associated with HLH. The condition has been described in both adults and children (Giard et al. 2016; Lin et al. 2016; Alonso et al. 2017). The presence of rampant immune activation and dysregulation with associated ALF makes the diagnosis of HLH difficult. Significant liver damage with ALF and HLH puts these children at premature risk of dying in the absence of treatment. Some of these children require liver transplantation. A retrospective study from Amir et al. evaluated children with HLH in association with ALF who required liver transplant. Six out of 9 patients remained alive at a median follow up of 24 months (Amir et al. 2016).
Diagnostic criteria for HLH
Clinical features
The diagnosis of HLH requires high index of suspicion. A timely diagnosis is therefore critical in order to start therapy before extensive damage by hypercytokinemia becomes irreversible. There is no specific clinical manifestation or a single laboratory finding that is pathognomonic for HLH. A triad of prolonged and unexplained fever, hepatosplenomegaly, and cytopenias should prompt investigation for HLH (Allen et al. 2008; Gupta and Weitzman 2010; Jordan et al. 2011; Weitzman 2011). Currently, the diagnosis is mostly based on a set of clinical and laboratory features. The diagnostic guideline for HLH was presented in 1991 (Henter et al. 1991b) and subsequently used by the Histiocyte Society in their HLH-94 study (Henter et al. 1997). These diagnostic criteria were later updated with few changes in the HLH-2004 study (Henter et al. 2007) (Tables 3 and 4). The initial bone marrow aspirate (BMA) and biopsy may not show any evidence of hemophagocytosis (Henter et al. 1991b). Lack or complete absence of hemophagocytosis on an initial BMA does not rule out the diagnosis of HLH and should not be a reason for delay in commencement on treatment (Gupta et al. 2008).
Table 3:

*
A higer ferritin levels >3000 μg/L is considered highly indicative of HLH.
Table 4:
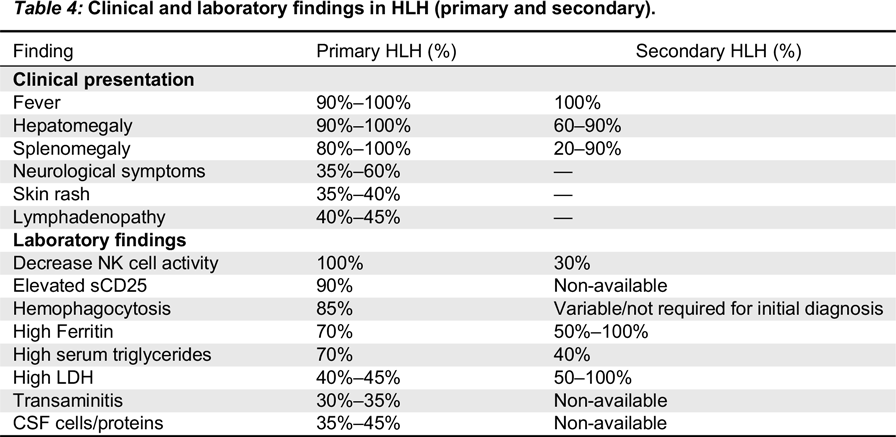
Although the HLH-2004 study mentions a ferritin level of >500 μg/L as one of the diagnostic criteria, it is generally considered that a level of >3000 μg/L should be concerning (Weitzman 2011), and a level >10 000 μg/L is highly suspicious for HLH diagnosis (Allen et al. 2008). CD25, a soluble IL-2 receptor (sIL-2r) is a marker of widespread lymphocyte activation and hyper-inflammation. An elevated sCD25 level, defined as >2 SD above the mean for age, is also one of the diagnostic criteria (Komp et al. 1989).
sCD-163 is a receptor for hemoglobin-haptoglobin complex, a marker for activation of alternative-pathway scavenger macrophages. Elevated levels of sCD-163 are seen in HLH that are higher than those associated with infections, autoimmune diseases, and cancer (Schaer et al. 2005). Recent studies have proposed the combination of sCD25 (produced by activated T cells and dendritic cells) and sCD163 (activated macrophages) as a useful tool in the diagnosis and surveillance of HLH disease activity (Weitzman 2011).
It is difficult to distinguish between primary and secondary HLH based on the currently used diagnostic criteria. It is important to rapidly diagnose familial cases so as to plan treatment in a timely manner. The only curative treatment at this time for familial HLH is HSCT. The molecular diagnostic testing takes a few weeks. In search of rapid diagnosis, Bryceson et al. published prospective evaluation of an NK cell and T cell degranulation assay done by flow cytometry. In cases where there is defective or abnormal resting NK cell degranulation, FHL 3–5 is likely. If the activated NK cell and cytotoxic T cell degranulation is abnormal, a familial HLH is likely. Patients with Chediak-Higashi and Griscelli syndrome type 2 also have defective NK cell degranulation (Bryceson et al. 2012).
Flow cytometry for perforin protein, SAP, and XIAP expression can also help as rapid screening tests for FHL2, XLP1, and XLP2, respectively. It is also important to take in consideration that flow cytometry screening can miss situations where the protein is present but not functional (Kogawa et al. 2002; Marsh et al. 2010a, 2010b; Johnson et al. 2011; Zhang et al. 2014).
Genetic testing is considered a gold standard in diagnosing mutations responsible for FHLH. Therefore, it is mandatory to investigate genetic mutations in a suspected case. Once the mutations are identified in the index patient, the siblings and parents should also be tested to establish the carrier status. This is necessary not only to find a potential stem cell donor, but for genetic counselling as well (Gholam et al. 2011).
Some of the tests, for example sIL2r and NK cell cytotoxicity, are only available in specialized laboratories. If the diagnosis of HLH is strongly suspected on the basis of clinical picture and the available tests, treatment should not be delayed as it can compromise outcome.
Central nervous system and HLH (CNS-HLH)
Neurological involvement is a common presentation in HLH, affecting overall outcome (Haddad et al. 1997; Ouachee-Chardin et al. 2006). About 10%–73% of children with HLH have associated increased cell count and (or) protein content in the cerebrospinal fluid (CSF), either at presentation or during the course of the disease (Kim et al. 2012). In 2008, the Histiocyte Society looked at the spectrum of CNS manifestations in 193 patients diagnosed with HLH and revealed neurological symptoms in 72 (37%) patients at diagnosis, abnormal CSF in 101 (52%), and either or both in 122 (63%). Moreover, late sequel and death were more frequent in the group with both abnormal CSF and neurological symptoms (Horne et al. 2008). Later, another Korean study found 23 of 50 patients with neurological signs had a relatively poor outcome compared to those with non CNS-HLH. Therefore, it is deemed important to investigate early in the course for CNS involvement in patients with HLH (Kim et al. 2012).
Treatment
Primary HLH without treatment is a rapidly fatal disease. In 1983, Janka reported 5% survival in a review of 121 cases (Janka 1983). Early diagnosis and treatment has improved outcome considerably. Introduction of epipodophyllotoxin derivatives has especially improved the remission status of these patients. Etoposide was first reported in 1980 as a treatment for HLH among 3 children (Ambruso et al. 1980). Plasma exchange was later attempted with transient resolution of symptoms in few patients (Ladisch et al. 1982). A combination therapy including etoposide, steroids, intrathecal methotrexate and cranial radiation therapy in 1985 revealed prolonged remission in patients with FHLH (Fischer et al. 1985). Henter et al. also reported prolonged remission with the combination therapy of teniposide in 7 children (Henter et al. 1986). It is now well understood that FHLH is associated with defective apoptotic mechanisms (Fadeel et al. 1999), and etoposide being very effective in initiating apoptosis, became the cornerstone of treatment in HLH (Henter et al. 1997). In murine FHL, etoposide acts selectively on activated T cells and controls immune dysregulation (Johnson et al. 2014). Similarly, the effect of corticosteroids can be explained by the anti-inflammatory and pro-apoptotic properties and cyclosporine A (CSA) in reducing T cell activity in HLH.
In 1994, the Histiocyte Society proposed a combination therapy comprising of etoposide, CSA and dexamethasone followed by HSCT for familial HLH. The regimen consisted of induction therapy with etoposide and dexamethasone, with gradual wean over 8 weeks followed by HSCT as soon as a suitable donor is available. In the absence of availability of a donor, continuation therapy was advised. Moreover, individuals with progressive CNS disease also received intrathecal methotrexate (Henter et al. 1997). The outcome of children treated as per the Histiocyte Society’s HLH-1994 protocol was published in 2011, showing a 5-year survival rate of 54% ± 6%. This was even better in children who underwent HSCT (66% ± 8%) (Trottestam et al. 2011). In their subsequent study (HLH-2004), the Histiocyte Society revised the HLH-94 protocol and added CSA upfront at the start of induction, instead of starting in the continuation phase, with an aim to get early remission. Recently, results of the HLH-2004 study have been published and have shown overall 62% (230/369) of patients were alive at the median follow up of 5.2 years. This study has also shown good results with the etoposide and dexamethasone combination therapy. However, there was no considerable improvement in overall outcome by adding upfront oral CSA (Bergsten et al. 2017).
On the other hand, a second regimen adopted for over 15 years in Europe, comprised of an induction phase that included rabbit anti-thymocyte globulin (rATG) and methyl-prednisone (with gradual wean). This is followed by a continuation therapy consisting of cyclosporine and intermittent prednisone pulses until HSCT. Individuals with CNS involvement received a combination of intrathecal methotrexate and corticosteroids, similar to the HLH-94 protocol. The complete response was achieved in 73%, with partial response in 24% of patients (Mahlaoui et al. 2007).
There are new therapeutic approaches to improve survival of patients with HLH. An innovative trial by a Cincinnati group, Hybrid Immunotherapy in HLH, was recently completed in children <18 years of age. It comprised of rATG, etoposide, dexamethasone, intrathecal methotrexate, and hydrocortisone with a belief that a combination of two proven induction regimens (from the Histiocyte Society and Mahlaoui et al.) will result in comparable or even better remission rates. The evaluation of this trial is currently in progress (clinicaltrials.gov NCT01104025) (Children’s Hospital Medical Center). Another clinical trial on tocilizumab (anti-IL-6 antibody) is underway. The excessive inflammation in HLH is cytokine driven, and standard modalities do not target cytokine storms directly in HLH. The aim of using Tocilizumab is to decrease cytokines in HLH (clinical trials.gov NCT02007239) (Philadelphia). Anti-Interferon Gamma Monoclonal Antibody, N1-0501 (Novimmune), a phase 2 study is underway in the United States and Europe to evaluate safety and efficacy of N1-0501 in primary HLH. Unpublished data on 13 patients have shown satisfactory response in patients who failed first line therapy (Jordan et al. 2015).
The first report of a successful HSCT in HLH was published in 1986 (Fischer et al. 1986). A study of 86 children who were treated on the HLH-94 protocol followed by HSCT between 1995–2000 showed overall estimated survival rate of 64% (Horne et al. 2005). In earlier reports, use of myeloablative conditioning regimens were associated with increased transplant related complications in the first 100 days post-transplant. This included veno-occlussive disease of the liver, respiratory complications, and overwhelming infections (Horne et al. 2005; Ouachee-Chardin et al. 2006). Subsequently, there has been increasing use of reduced intensity conditioning (RIC) regimens, which have improved the outcome in these patients. In 2010, Marsh et al. published their experience in 26 patients, and found 3-year probability of survival when treated with RIC (alemtuzumab, fludarabin and melphalan based regimen) at 92% (Marsh et al. 2011). Comparable but slightly lower survival rates of 85% at 3-year median follow up in 25 patients treated with RIC was also reported by Cooper et al. (Cooper et al. 2008).
Despite advances in management and treatment of HLH, approximately 25%–50% patients fail to achieve remission with conventional treatment, including HSCT, and require salvage therapy. Many agents have been used in HLH including rituximab, rATG, and alemtuzumab (Mahlaoui et al. 2007; Strout et al. 2010; Chellapandian et al. 2013; Marsh et al. 2013b). Rituximab, a monoclonal CD20-antibody has been used in patients with EBV-associated HLH with fairly well response. A retrospective study of 42 patients with EBV associated HLH, treated with rituximab in combination with conventional HLH-directed therapy, showed improved clinical and laboratory parameters (Chellapandian et al. 2013). Similarly, when HLH becomes refractory to various immunosuppressive agents, it requires suppression of immune response at different levels, including cytotoxic CD8+ T cells, NK cells, cytokine producing CD4+ T cells, and antigen presenting cells. CD52 antigen is a small GPI-anchored protein expressed on lymphocytes, monocytes, macrophages, and dendritic cells. Alemtuzumab is a monoclonal antibody targeting CD52 antigen. Alemtuzumab has been reported as a bridging therapy to HSCT in 1 patient with refractory HLH (Strout et al. 2010). A study by Marsh et al. involving 22 pediatric and adult patients treated with alemtuzumab experienced noticeable improvement in clinical symptoms and laboratory markers, and 77% survived to undergo allogeneic HSCT (Marsh et al. 2013a).
Conclusion
HLH is a life threatening hyper-inflammatory syndrome if left untreated. Early diagnosis and prompt initiation of treatment is a key to improve survival. Over the decades, with increasing awareness of this condition amongst caregivers, better understanding of pathophysiology, use of consensus treatment protocols, and with changes in the stem cell conditioning, the overall outcome of patients has significantly improved, but there is still room for much improvement. There is a need of better diagnostic tools and innovative up front and salvage therapy.
REFERENCES
Allen C.E., Yu X., Kozinetz C.A., and McClain K.L. 2008. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer. 50:1227–1235.
Alonso E.M., Horslen S.P., Behrens E.M., and Doo E. 2017. Pediatric acute liver failure of undetermined cause: A research workshop. Hepatology. 65:1026–1037.
Ambruso D.R., Hays T., Zwartjes W.J., Tubergen D.G., and Favara B.E. 1980. Successful treatment of lymphohistiocytic reticulosis with phagocytosis with epipodophyllotoxin VP 16-213. Cancer. 45:2516–2520.
Amir A.Z., Ling S.C., Naqvi A., Weitzman S., Fecteau A., Grant D., Ghanekar A., Cattral M., Nalli N., Cutz E., Kamath B., Jones N., de Angelis M., Ng V., and Avitzur Y. 2016. Liver transplantation for children with acute liver failure associated with secondary hemophagocytic lymphohistiocytosis. Liver Transpl. 22:1245–1253.
Aricò M., Janka G., Fischer A., Henter J.I., Blanche S., Elinder G., Martinetti M., and Rusca M.P. 1996. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 10:197–203.
Asano T., Kogawa K., Morimoto A., Ishida Y., Suzuki N., Ohga S., Kudo K., Ohta S., Wakiguchi H., Tabuchi K., Kato S., and Ishii E. 2012. Hemophagocytic lymphohistiocytosis after hematopoietic stem cell transplantation in children: A nationwide survey in Japan. Pediatr. Blood Cancer. 59:110–114.
Bergsten E., Horne A., Arico M., Astigarraga I., Egeler R.M., Filipovich A.H., Ishii E., Janka G., Ladisch S., Lehmberg K., McClain K.L., Minkov M., Montgomery S., Nanduri V., Rosso D., and Henter J.I. 2017. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: Long-term results of the cooperative HLH-2004 study. Blood. 130:2728–2738.
Bode S.F.Ammann S.Al-Herz W.Bataneant M.Dvorak C.C.Gehring S.Gennery A.Gilmour K.C.Gonzalez-Granado L.I.Gross-Wieltsch U.Ifversen M.Lingman-Framme J.Matthes-Martin S.Mesters R.Meyts I.van Montfrans J.M.Pachlopnik Schmid J.Pai S.Y.Soler-Palacin P.Schuermann U.Schuster V.Seidel M.G.Speckmann C.Stepensky P.Sykora K.W.Tesi B.Vraetz T.Waruiru C.Bryceson Y.T.Moshous D.Lehmberg K.Jordan M.B.Ehl S., and Inborn Errors Working Party of the EBMT. 2015. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: Implications for differential diagnosis and pathogenesis. Haematologica. 100:978–988.
Brisse E., Wouters C.H., and Matthys P. 2015. Hemophagocytic lymphohistiocytosis (HLH): A heterogeneous spectrum of cytokine-driven immune disorders. Cytokine Growth Factor Rev. 26:263–280.
Bryceson Y.T., Pende D., Maul-Pavicic A., Gilmour K.C., Ufheil H., Vraetz T., Chiang S.C., Marcenaro S., Meazza R., Bondzio I., Walshe D., Janka G., Lehmberg K., Beutel K., Zur Stadt U., Binder N., Arico M., Moretta L., Henter J.I., and Ehl S. 2012. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 119:2754–2763.
Chellapandian D., Das R., Zelley K., Wiener S.J., Zhao H., Teachey D.T., Nichols K.E., and Group E.-H.R.S. 2013. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. 162:376–382.
Children’s Hospital Medical Center, C. Hybrid Immunotherapy for Hemophagocytic Lymphohistiocytosis. In Clincal Trial.gov NCT01104025. Available at: https://clinicaltrials.gov/ct2/show/NCT01104025 [accessed 13 March 2018]
Cooper N., Rao K., Goulden N., Webb D., Amrolia P., and Veys P. 2008. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and langerhans cell histiocytosis. Bone Marrow Transplant. 42(Suppl 2):S47–S50.
Côte M., Menager M.M., Burgess A., et al. 2009. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J. Clin. Invest. 119:3765–3773.
Danielian S., Basile N., Rocco C., Prieto E., Rossi J., Barsotti D., Roche P.A., Bernasconi A., Oleastro M., Zelazko M., and Braier J. 2010. Novel syntaxin 11 gene (STX11) mutation in three Argentinean patients with hemophagocytic lymphohistiocytosis. J. Clin. Immunol. 30:330–337.
Degar B. 2015. Familial hemophagocytic lymphohistiocytosis. Hematol. Oncol. Clin. North Am. 29:903–913.
Fadeel B., Orrenius S., and Henter J.I. 1999. Induction of apoptosis and caspase activation in cells obtained from familial haemophagocytic lymphohistiocytosis patients. Br. J. Haematol. 106:406–415.
Faitelson Y. and Grunebaum E. 2014. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders. Clin Immunol. 155:118–125.
Falini B., Pileri S., De Solas I., Martelli M.F., Mason D.Y., Delsol G., Gatter K.C., and Fagioli M. 1990. Peripheral T-cell lymphoma associated with hemophagocytic syndrome. Blood. 75:434–444.
Farquhar J.W. and Claireaux A.E. 1952. Familial hemophagoctic reticulosis. Arch. Dis. Child. 27:519–525.
Farquhar J.W., Macgregor A.R., and Richmond J., 1958. Familial haemophagocytic reticulosis. Br. Med. J. 2:1561–1564.
Feldmann J., Callebaut I., Raposo G., Certain S., Bacq D., Dumont C., Lambert N., Ouachee-Chardin M., Chedeville G., Tamary H., Minard-Colin V., Vilmer E., Blanche S., le Deist F., Fischer A., and de Saint Basile G. 2003. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 115:461–473.
Fischer A., Virelizier J.L., Arenzana-Seisdedos F., Perez N., Nezelof C., and Griscelli C. 1985. Treatment of four patients with erythrophagocytic lymphohistiocytosis by a combination of epipodophyllotoxin, steroids, intrathecal methotrexate and cranial irradiation. Pediatrics. 76:263–68.
Fischer A., Cerf-Bensussan N., Blanche S., Le Deist F., Bremard-Oury C., Leverger G., Schaison G., Durandy A., and Griscelli C. 1986. Allogeneic bone marrow transplantation for erythrophagocytic lymphohistiocytosis. J. Pediatr. 108:267–270.
George M.R. 2014. Hemophagocytic lymphohistiocytosis: Review of etiologies and management. J. Blood Med. 5:69–86.
Gholam C., Grigoriadou S., Gilmour K.C., and Gaspar H.B. 2011. Familial haemophagocytic lymphohistiocytosis: Advances in the genetic basis, diagnosis and management. Clin. Exp. Immunol. 163:271–283.
Ghosh S., Bienemann K., Boztug K., and Borkhardt A. 2014. Interlukin-2-inducible T-cell kinase (ITK) deficiency—clinical and molecular aspects. J. Clin. Immunol. 34:892–899.
Giard J.M., Decker K.A., Lai J.C., Gill R.M., Logan A.C., and Fix O.K. 2016. Acute liver failure secondary to hemophagocytic lymphohistiocytosis during pregnancy. ACG Case Rep. J. 3:162–164.
Grom A.A. 2017. Primary hemophagocytic lymphohistiocytosis and macrophage activation syndrome: The importance of timely clinical differentiation. J. Pediatr. 189:19–21.e1.
Grom A.A. and Mellins E.D. 2010. Macrophage activation syndrome: Advances towards understanding pathogenesis. Curr. Opin. Rheumatol. 22:561–566.
Gupta S. and Weitzman S. 2010. Primary and secondary hemophagocytic lymphohistiocytosis: Clinical features, pathogenesis and therapy. Expert Rev. Clin. Immunol. 6:137–154.
Gupta A., Tyrrell P., Valani R., Benseler S., Weitzman S., and Abdelhaleem M. 2008. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer. 51:402–404.
Gürgey A., Göğüş S., Ozyürek E., Aslan D., Gümrük F., Cetin M., Yüce A., Ceyhan M., Seçmeer G., Yetgin S., and Hiçsönmez G. 2003. Primary hemophagocytic lymphohistiocytosis in Turkish children. Pediatr. Hematol. Oncol. 20:367–371.
Haddad E., Sulis M.L., Jabado N., Blanche S., Fischer A., and Tardieu M. 1997. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood. 89:794–800.
Henter J.I., Elinder G., Finkel Y., and Soder O. 1986. Successful induction with chemotherapy including teniposide in familial erythrophagocytic lymphohistiocytosis. Lancet. 328:1402.
Henter J.I., Elinder G., Söder O., and Öst Å. 1991a. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Pædiatrica. 80:428–435.
Henter J.I., Elinder G., and Ost A. 1991b. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin. Oncol. 18:29–33.
Henter J.I., Elinder G., Söder O., Hansson M., Andersson B., and Andersson U. 1991c. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 78:2918–2922.
Henter J.I., Aricò M., Egeler R.M., Elinder G., Favara B.E., Filipovich A.H., Gadner H., Imashuku S., Janka-Schaub G., Komp D., Ladisch S., and Webb D. 1997. HLH-94: A treatment protocol for hemophagocytic lymphohistiocytosis. Med. Pediatr. Oncol. 28:342–347.
Henter J.I., Horne A., Arico M., Egeler R.M., Filipovich A.H., Imashuku S., Ladisch S., Mcclain K., Webb D., Winiarski J., and Janka G. 2007. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer. 48:124–131.
Horne A.Janka G.Maarten Egeler R.Gadner H.Imashuku S.Ladisch S.Locatelli F.Montgomery S.M.Webb D.Winiarski J.Filipovich A.H.Henter J.I., and Histiocyte Society. 2005. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br. J. Haematol. 129:622–630.
Horne A., Trottestam H., Arico M., Egeler R.M., Filipovich A.H., Gadner H., Imashuku S., Ladisch S., Webb D., Janka G., Henter J.I., and Histiocyte S. 2008. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br. J. Haematol. 140:327–335.
Ishii E., Ohga S., Tanimura M., Imashuku S., Sako M., Mizutani S., and Miyazaki S. 1998. Clinical and epidemiologic studies of familial hemophagocytic lymphohistiocytosis in Japan. Japan LCH Study Group. Med. Pediatr. Oncol. 30:276–283.
Ishii E., Ohga S., Imashuku S., Yasukawa M., Tsuda H., Miura I., Yamamoto K., Horiuchi H., Takada K., Ohshima K., Nakamura S., Kinukawa N., Oshimi K., and Kawa K. 2007. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int. J. Hematol. 86:58–65.
Janka G., Imashuku S., Elinder G., Schneider M., and Henter J.I. 1998. Infection and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol. Oncol. Clin. North Am. 12:435–444.
Janka G.E. 1983. Familial hemophagocytic lymphohistiocytosis. Eur. J. Pediatr. 140:221–230.
Janka G.E. 2007a. Familial and acquired hemophagocytic lymphohistiocytosis. Eur. J. Pediatr. 166:95–109.
Janka G.E. 2007b. Hemophagocytic syndromes. Blood Rev. 21:245–253.
Janka G.E. 2012. Familial and acquired hemophagocytic lymphohistiocytosis. Annu. Rev. Med. 63:233–246.
Janka G.E. and Lehmberg K. 2014. Hemophagocytic syndromes—an update. Blood Rev. 28:135–142.
Johnson T.S., Villanueva J., Filipovich A.H., Marsh R.A., and Bleesing J.J. 2011. Contemporary diagnostic methods for hemophagocytic lymphohistiocytic disorders. J. Immunol. Methods. 364:1–13.
Johnson T.S., Terrell C.E., Millen S.H., Katz J.D., Hildeman D.A., and Jordan M.B. 2014. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J. Immunol. 192:84–91.
Jordan M.B., Allen C.E., Weitzman S., Filipovich A.H., and Mcclain K.L. 2011. How I treat hemophagocytic lymphohistiocytosis. Blood. 118:4041–4052.
Jordan M.B.Locatelli F.Allen C.De Benedetti F.Grom A.Ballabio M.Ferlin W.De Min C., and on behalf of the NI-0501-04 Study Group. 2015. A novel targeted approach to the treatment of hemophagocytic lymphohistiocytosis (HLH) with an anti-interferon gamma (IFNy) monoclonal antibody (mAb), N1-0501: first results from a pilot phase 2 study in children with primary HLH. Blood. 126:LBA-3 (abstract).
Kaufman K.M., Linghu B., Szustakowski J.D., Husami A., Yang F., Zhang K., Filipovich A.H., Fall N., Harley J.B., Nirmala N.R., and Grom A.A. 2014. Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 66:3486–3495.
Kim H.K., Kim H.G., Cho S.J., Hong Y.M., Sohn S., Yoo E.S., Chung W.S., and Ryu K.H. 2011. Clinical characteristics of hemophagocytic lymphohistiocytosis related to Kawasaki disease. Pediatr. Hematol. Oncol. 28:230–236.
Kim M.M., Yum M.S., Choi H.W., Ko T.S., Im H.J., Seo J.J., and Koh K.N. 2012. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J. Hematol. 47:273–280.
Kogawa K., Lee S.M., Villanueva J., Marmer D., Sumegi J., and Filipovich A.H. 2002. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood. 99:61–66.
Komp D.M., McNamara J., and Buckley P. 1989. Elevated soluble interleukin-2 receptor in childhood hemophagocytic histiocytic syndromes. Blood. 73:2128–2132.
Ladisch S., Ho W., Matheson D., Pilkington R., and Hartman G. 1982. Immunologic and clinical effects of repeated blood exchange in familial erythrophagocytic lymphohistiocytosis. Blood. 60:814–821.
Lin S., Li Y., Long J., Liu Q., Yang F., and He Y. 2016. Acute liver failure caused by hemophagocytic lymphohistiocytosis in adults: A case report and review of the literature. Medicine (Baltimore). 95:e5431.
Macartney C.A., Weitzman S., Wood S.M., Bansal D., Steele M., Meeths M., Abdelhaleem M., and Bryceson Y.T. 2011. Unusual functional manifestations of a novel STX11 frameshift mutation in two infants with familial hemophagocytic lymphohistiocytosis type 4 (FHL4). Pediatr. Blood Cancer. 56:654–657.
Madkaikar M., Shabrish S., and Desai M. 2016. Current updates on classification, diagnosis and treatment of hemophagocytic lymphohistiocytosis (HLH). Indian J. Pediatr. 83:434–443.
Mahlaoui N., Ouachee-Chardin M., de Saint Basile G., Neven B., Picard C., Blanche S., and Fischer A. 2007. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: A single-center retrospective report of 38 patients. Pediatrics. 120:e622–e628.
Marsh R.A., Bleesing J.J., and Filipovich A.H. 2010a. Using flow cytometry to screen patients for X-linked lymphoproliferative disease due to SAP deficiency and Xiap deficiency. J. Immunol. Methods. 362:1–9.
Marsh R.A., Satake N., Biroschak J., Jacobs T., Johnson J., Jordan M.B., Bleesing J.J., Filipovich A.H., and Zhang K. 2010b. STX11 mutations and clinical phenotypes of familial hemophagocytic lymphohistiocytosis in North America. Pediatr. Blood Cancer. 55:134–40.
Marsh R.A., Jordan M.B., and Filipovich A.H. 2011. Reduced-intensity conditioning haematopoietic cell transplantation for haemophagocytic lymphohistiocytosis: An important step forward. Br. J. Haematol. 154:556–563.
Marsh R.A., Allen C.E., Mcclain K.L., Weinstein J.L., Kanter J., Skiles J., Lee N.D., Khan S.P., Lawrence J., Mo J.Q., Bleesing J.J., Filipovich A.H., and Jordan M.B. 2013a. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr. Blood Cancer. 60:101–109.
Marsh R.A., Kim M.O., Liu C., Bellman D., Hart L., Grimley M., Kumar A., Jodele S., Myers K.C., Chandra S., Leemhuis T., Mehta P.A., Bleesing J.J., Davies S.M., Jordan M.B., and Filipovich A.H. 2013b. An intermediate alemtuzumab schedule reduces the incidence of mixed chimerism following reduced-intensity conditioning hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Biol. Blood Marrow Transplant. 19:1625–1631.
Moritake H., Kamimura S., Nunoi H., Nakayama H., Suminoe A., Inada H., Inagaki J., Yanai F., Okamoto Y., Shinkoda Y., Shimomura M., Itonaga N., Hotta N., Hidaka Y., Ohara O., Yanagimachi M., Nakajima N., Okamura J., and Kawano Y. 2014. Clinical characteristics and genetic analysis of childhood acute lymphoblastic leukemia with hemophagocytic lymphohistiocytosis: A Japanese retrospective study by the Kyushu-Yamaguchi Children’s Cancer Study Group. Int. J. Hematol. 100:70–78.
Murphy E.P., Mo J., and Yoon J.M. 2015. Secondary hemophagocytic lymphohistiocytosis in a patient with favorable histology wilms tumor. J. Pediatr. Hematol. Oncol. 37:e494–e496.
Nagai K., Yamamoto K., Fujiwara H., An J., Ochi T., Suemori K., Yasumi T., Tauchi H., Koh K., Sato M., Morimoto A., Heike T., Ishii E., and Yasukawa M. 2010. Subtypes of familial hemophagocytic lymphohistiocytosis in Japan based on genetic and functional analyses of cytotoxic T lymphocytes. PLoS ONE. 5:e14173.
Niece J.A., Rogers Z.R., Ahmad N., Langevin A.M., and McClain K.L. 2010. Hemophagocytic lymphohistiocytosis in Texas: Observations on ethinicity and race. Pediatr. Blood Cancer. 54:424–428.
Ohadi M., Lalloz M.R., Sham P., Zhao J., Dearlove A.M., Shiach C., Kinsey S., Rhodes M., and Layton D.M. 1999. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am. J. Hum. Genet. 64:165–171.
Ouachee-Chardin M., Elie C., de Saint Basile G., le Deist F., Mahlaoui N., Picard C., Neven B., Casanova J.L., Tardieu M., Cavazzana-Calvo M., Blanche S., and Fischer A. 2006. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: A single-center report of 48 patients. Pediatrics. 117:e743–e750.
Parvaneh N., Filipovich A.H., and Borkhardt A. 2013. Primary immunodeficiencies predisposed to Epstein-Barr virus-driven haematological diseases. Br. J. Haematol. 162:573–586.
Philadelphia, C.S.H.O. Cytokine blockade with tocilizumab in patients with cytokine release syndrome and hemophagocytic lymhohistiocytosis. Cancer trials.gov. NCT 02007239. Available at: https://clinicaltrials.gov/ct2/show/NCT02007239 [accessed 13 March 2018].
Ramos-Casals M., Brito-Zerón P., López-Guillermo A., Khamashta M.A., and Bosch X. 2014. Adult haemophagocytic syndrome. Lancet. 383:1503–1516.
Risdall R.J., McKenna R.W., Nesbit M.E., Krivit W., Balfour H.H. Jr., Simmons R.L., and Brunning R.D. 1979. Virus-associated hemophagocytic syndrome: A benign histiocytic proliferation distinct from malignant histiocytosis. Cancer. 44:993–1002.
Rosado F.G. and Kim A.S. 2013. Hemophagocytic lymphohistiocytosis: An update on diagnosis and pathogenesis. Am. J. Clin. Pathol. 139:713–727.
Rouphael N.G., Talati N.J., Vaughan C., Cunningham K., Moreira R., and Gould C. 2007. Infections associated with haemophagocytic syndrome. The Lancet Infect. Dis. 7:814–822.
Schaer D.J., Schleiffenbaum B., Kurrer M., Imhof A., Bächli E., Fehr J., Moller H.J., Moestrup S.K., and Schaffner A. 2005. Soluble hemoglobin-haptoglobin scavenger receptor CD163 as a lineage-specific marker in the reactive hemophagocytic syndrome. Eur. J. Haematol. 74:6–10.
Scott R.B. and Robb-Smith A.H.T. 1939. Histiocytic medullary histiocytosis. Lancet. 234:194–198.
Stepp S.E., Dufourcq-Lagelouse R., Le Deist F., Bhawan S., Certain S., Mathew P.A., Henter J.I., Bennett M., Fischer A., de Saint Basile G., and Kumar V. 1999. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 286:1957–1959.
Strout M.P., Seropian S., and Berliner N. 2010. Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic lymphohistiocytosis. Nat. Rev. Clin. Oncol. 7:415–420.
Trottestam H., Horne A., Arico M., Egeler R.M., Filipovich A.H., Gadner H., Imashuku S., Ladisch S., Webb D., Janka G., Henter J.I., and Histiocyte S. 2011. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: Long-term results of the HLH-94 treatment protocol. Blood. 118:4577–4584.
Usmani G.N., Woda B.A., and Newburger P.E. 2013. Advances in understanding the pathogenesis of HLH. Br. J. Haematol. 161:609–622.
Weitzman S. 2011. Approach to hemophagocytic syndromes. Hematology Am. Soc. Hematol. Educ. Program. 2011:178–83.
Wu J.R., Yuan L.X., Ma Z.G., Chen X.X., Gu L., and Gao J. 2013. GDF15-mediated upregulation of ferroportin plays a key role in the development of hyperferritinemia in children with hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer. 60:940–945.
Yang X., Wada T., Imadome K., Nishida N., Mukai T., Fujiwara M., Kawashima H., Kato F., Fujiwara S., Yachie A., Zhao X., Miyawaki T., and Kanegane H. 2012. Characterization of Epstein-Barr virus (EBV)-infected cells in EBV-associated hemophagocytic lymphohistiocytosis in two patients with X-linked lymphoproliferative syndrome type 1 and type 2. Herpesviridae. 3:1.
Yang S.L., Xu X.J, Tang Y.M., Song H., Xu W.Q., Zhao F.Y., and Shen D.Y. 2016. Associations between inflammatory cytokines and organ damage in pediatric patients with hemophagocytic lymphohistioctosis. Cytokine. 85:14–17.
Yoon H.S., Kim H.J., Yoo K.H., Sung K.W., Koo H.H., Kang H.J., Shin H.Y., Ahn H.S., Kim J.Y., Lim Y.T., Bae K.W., Lee K.O., Shin J.S., Lee S.T., Chung H.S., Kim S.H., Park C.J., Chi H.S., Im H.J., and Seo J.J. 2010. UNC13D is the predominant causative gene with recurrent splicing mutations in Korean patients with familial hemophagocytic lymphohistiocytosis. Haematologica. 95:622–626.
Zhang K., Chandrakasan S., Chapman H., Valencia C.A., Husami A., Kissell D., Johnson J. A., and Filipovich A. H. 2014. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood. 124:1331–1334.
Zheng F., Li J., Zha H., Zhang J., Zhang Z., and Cheng F. 2016. ITK gene mutation: Effect on survival of children with severe hemophagocytic lymphohistiocytosis. Indian J. Pediatr. 83(11):1349–1352.
zur Stadt U., Schmidt S., Kasper B., Beutel K., Diler A.S., Henter J.I., Kabisch H., Schneppenheim R., Nürnberg P., Janka G., and Hennies H.C. 2005. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum. Mol. Genet. 14:827–834.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 5 • Number 1 • March 2018
Pages: 1 - 15
History
Received: 4 September 2017
Accepted: 31 January 2018
Accepted manuscript online: 2 February 2018
Copyright
© 2018.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
SaimaAlvi and AhmedNaqvi. 2018. Advances in the diagnosis and management of haemophagocytic lymphohistiocytosis: a review of literature. LymphoSign Journal.
5(1): 1-15. https://doi.org/10.14785/lymphosign-2017-0010
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
