A novel mutation in Roifman syndrome redefines the boundaries of the Sm protein-binding site
Abstract
Roifman syndrome is an association of humoral immunodeficiency, growth retardation, spondyloepiphyseal dysplasia, developmental delay, retinal dystrophy, and unique dysmorphism. Compound heterozygote mutations in the RNU4ATAC gene, an essential component of the minor spliceosome, were found to be the culprit for this disorder. Here we report a novel mutation in the RNU4ATAC gene, involving position 116. This mutation redefines the Sm protein binding site of this gene.
Statement of novelty: We report here a patient with a novel mutation in the Sm protein-binding site that redefined its boundaries to include position 116.
Introduction
Roifman syndrome (OMIM #616651) was first identified in 4 males from 3 different families of diverse genetic backgrounds as a novel association of humoral immunodeficiency, pre- and postnatal growth retardation, spondyloepiphyseal dysplasia, developmental delay, retinal dystrophy, and unique facial dysmorphic features (Roifman 1997, 1999). Later, additional features were identified in these patients, including cardiac noncompaction (Mandel et al. 2001), hypogonadotrophic hypogonadism (Robertson et al. 2000), partial agenesis of the corpus callosum, hypocampal hypoplasia (Fairchild et al. 2011), and renal tubular dysfunction (de Vries et al. 2006).
Recently, rare compound heterozygote mutations in the RNU4ATAC gene were found to be the culprit for this disorder (Merico et al. 2015). This gene encodes for U4atac small nuclear RNA (snRNA), an essential component of the minor spliceosome, which is crucial for the correct splicing of about 800 genes carrying minor introns. The structural elements of the U4atac snRNA (Figure 1) include 2 elements named stem I and stem II, which base pair the U6atac required to form the catalytically active minor spliceosome. Stem I and stem II are separated by a 5′ stem-loop. Another stem-loop, the 3′ stem-loop, is followed by a sequence acting as a binding site for the Sm proteins, required for the assembly of the complex and its import into the nucleus. Roifman syndrome casual variants reported to date present a characteristic compound heterozygosity pattern with 1 variant involving the 5′ stem-loop or the Sm protein-binding site, whereas the other variant is in the stem II element—a newly implicated yet highly conserved element of the gene.
Figure 1:

Methods
Flow cytometry
Peripheral blood mononuclear cells were obtained by Ficoll–Hypaque density gradient centrifugation and surface phenotypes determined by flow cytometry on a Coulter EPICS V low cytometer (Beckman Coulter; Brea, CA, USA). All fluorescent labeled antibodies were purchased from eBiosciences (San Diego, CA, USA).
Serum concentration of immunoglobulin and specific antibodies
Serum concentrations of immunoglobulins were measured by nephelometry. Levels of serum antibodies to tetanus were measured by ELISA.
T- and B-cell proliferative response
Lymphocyte proliferative responses to mitogens including phytohemagglutinin (PHA) and anti-CD3 antibodies were determined by thymidine incorporation. All assays were performed in triplicate and were compared with simultaneously stimulated normal controls.
Sequencing analysis
The patient’s genomic DNA was extracted from peripheral blood lymphocytes using the Geneaid Genomic DNA Mini Kit. Genomic DNA was amplified by PCR with specific primers designed upstream and downstream of the RNU4ATAC gene. Sequencing was done using GenomeLab Dye Terminator Cycle Sequencing Quick Start Kit (Beckman Coulter) and analyzed on CEQ 8000 Genetic Analysis System (Beckman Coulter).
Case description
The patient is a 12-year-old male of Tamil descent, born at term with a low birth weight of 1.8 kg. He first presented at 11 weeks of age with a severe bilateral pneumonia with a complicated course requiring intubation and high flow ventilation. He then continued to suffer from recurrent pneumoniae as well as a reactive airway disease, and was treated with multiple courses of antibiotics, inhaled beta agonists, inhaled corticosteroids, and leukotriene receptor antagonists.
In addition to his respiratory disease, the patient suffered from global developmental delay. He sat at the age of 11 months and was able to walk and say “mom” and “dad” at the age of 2 years. He currently attends school with the assistance of a tutor. A brain MRI revealed mild ventriculomegaly and prominent extra-axial CSF spaces. He was noticed to have dysmorphic features, including short stature (height below the 3rd percentile for age, weight at the 50th percentile for age), brachydactyly, microcephaly (head circumference below the 3rd percentile), and hypoplastic shallow mid facies with a long philtrum and a thin upper lip (Figure 2). An initial skeletal survey at the age of 5 months revealed failure of ossification of the femoral heads and distal femora and proximal tibiae. A subsequent skeletal survey at 6.5 years of age was grossly abnormal: skeletal maturation was significantly advanced and there was a marked discrepancy between the bone age in the distal (10 years) versus the proximal (5 years) hand. There were diffuse epiphyseal and metaphyseal changes including small, flattened, and slightly broadened femoral heads (Figure 3). The distal tibiae and radial epiphyses were small and wedged (Figure 4), and the metacarpal heads were squared and flattened distally. Metaphyseal changes included irregular and slightly broadened proximal femoral metaphysis as well as prominent changes in the tibiae and distal femora bilaterally (Figure 5).
Figure 2:

Figure 3:
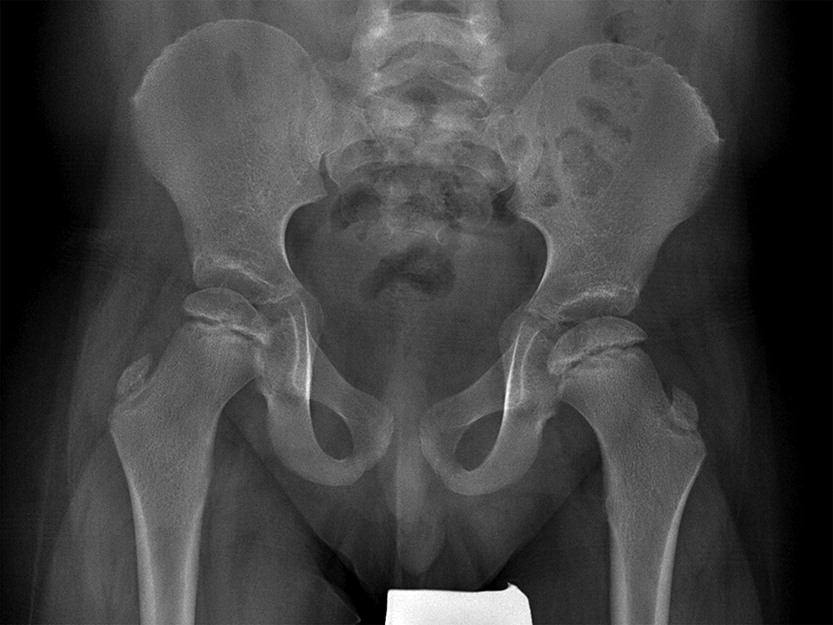
Figure 4:
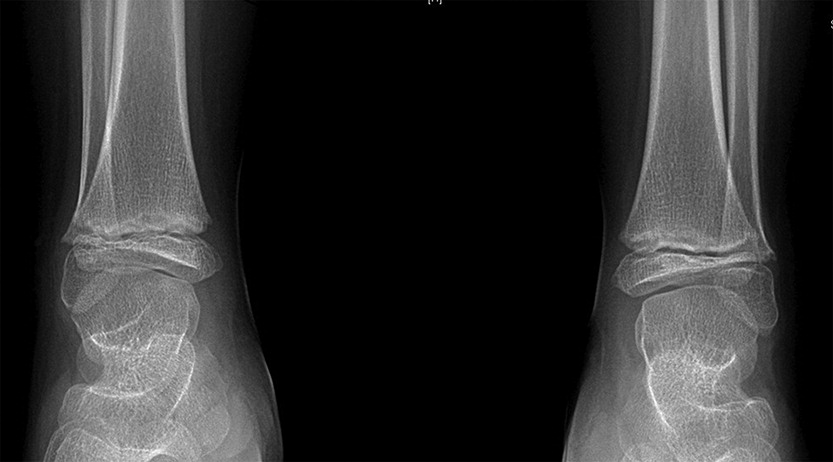
Figure 5:

At the age of 4 years, the patient started having poor night vision and decreased field of vision. At the age of 7 years, he was found to have complex retinal dystrophy with cystoid macular edema. He improved clinically on Brinzoamide drops.
The parents are nonconsanguineous and there is no history of primary immune deficiency in the family. The patient has an older, healthy sister and the mother has a history of 4 miscarriages.
An initial immunologic evaluation was significant for lymphopenia (1.53 × 109 cells/L) with significantly reduced CD19 cells (47 cells/µL). Numbers of all other lymphocyte markers were normal (CD3 count of 3417 cells/µL, CD4 count of 2604 cells/µL, CD8 count of 837 cells/µL, and NK cell count of 387 cells/µL). He had low immunoglobulin levels (IgG of 1.8 g/L, IgA of less than 0.1 g/L, and IgM of 0.2 g/L). T-cell mitogen response was normal with a stimulation index of 1529.3 in response to PHA.
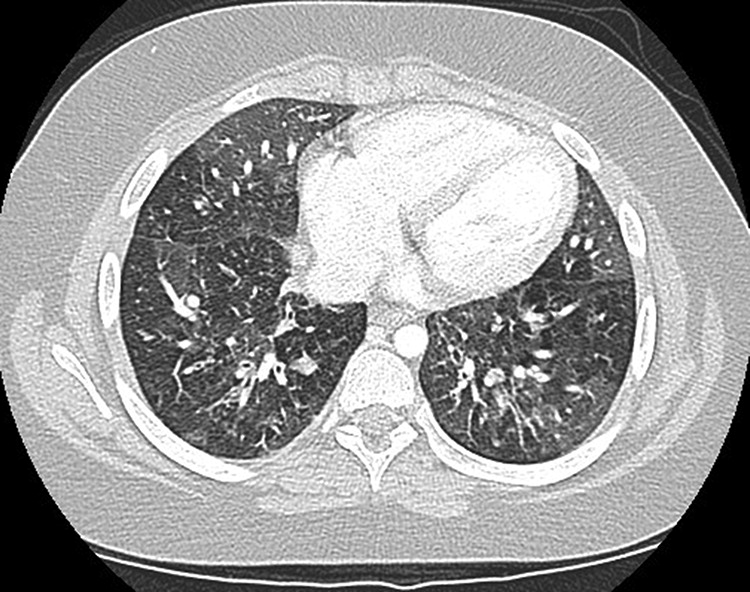
The patient’s immunoglobulin levels normalized over the course of the next year and his B-cell counts were variable. He was, however, unable to mount a long-standing response to tetanus and pneumococcal vaccines and had nonreactive titres for measles, mumps, rubella, and varicella despite appropriate vaccination. He was therefore started on IVIG treatment at the age of 11 years. Despite this treatment, the patient’s respiratory status continued to deteriorate and he eventually developed bronchiolitis obliterans following adenoviral infection (Figure 6). He was treated with 3 courses of pulse steroids followed by a 7-month course of daily oral steroids. Despite developing multiple complications of steroid treatment, the patient’s response to treatment was inadequate and he remains significantly debilitated from the respiratory standpoint, requiring constant oxygen support.
Figure 6:
Analysis of the RNU4ATAC gene revealed compound heterozygous mutation with a 116A>G mutation in 3′ stem loop domain and a 17G>A mutation in the Stem II domain. Although this mutation has not been previously reported, mutations in the same domains have been reported to be the cause of Roifman syndrome (Merico et al. 2015). The mother was found to be a carrier of the 17G>A mutation and the father of the 116A>G mutation.
Discussion
We report a patient with a novel mutation in the RNU4ATAC gene causing Roifman syndrome. The association between compound heterozygous mutations in the RNU4ATAC gene and Roifman syndrome was recently described (Merico et al. 2015). This gene encodes for a snRNA gene which is essential for minor intron splicing. As such, it is involved with the correct splicing of approximately 800 genes. Using RNA-seq methods, splicing changes were observed that included intron retention specific for minor (but not major) intron genes. Of these, 30 genes were identified whose pathogenesis is relevant to Roifman syndrome (developmental, neurocognitive, immune, skeletal, and retinal).
RNU4ATAC homozygous and compound heterozygous variants were also reported to cause MOPD1 (OMIM 210710) (Edery et al. 2011; He et al. 2011). MOPD1 (microcephalic osteodyslastic primordial dwarfism type I) is clinically distinguishable from Roifman syndrome, and is associated with pre- and postnatal lethality; severe prenatal microcephaly and brain malformations; intractable epilepsy; short and bowed limbs; absent or sparse hair; neuroendocrine dysfunction and distinct facial features including proptotic eyes, prominent nose or downturned nasal tip, micrognathia; and occasionally, skin and retinal hypopigmentation. Most MOPD1 causal variants are clustered in the 5′ stem-loop, while there are a few mutations in the Sm protein binding site, 3′ stem-loop or connecting the single strand region to stem I. In contrast, all Roifman syndrome patients described to date carry a compound heterozygous mutation with 1 variant in the 5′ stem loop critical region or the Sm protein binding site, while the other variant is in highly conserved positions in the stem II region, which is unique to Roifman syndrome.
Our patient presented with a similar phenotype to that previously reported, including IUGR, dysgammaglobulinemia, developmental delay, spondyloepiphyseal dysplasia, and distinctive dysmorphism, including microcephaly and hypoplastic shallow mid facies with a long philtrum and a thin upper lip (Figure 2). In contrast with previous reports, he did not have eosinophilia, elevated IgE, or eczema. He did, however, suffer from reactive airway disease and developed a significant chronic lung injury.
We present a patient with a novel compound heterozygous mutation in the RNU4ATAC gene. The 116A>G mutation is located at the edge of the 3′ stem-loop domain in an element which is considered of variable importance for splicing. This newly discovered variant (Shukla et al. 2002) led to a revised Sm protein binding site to include position 116 (Jafarifar et al. 2014). This is supported by a previous report (Raker et al. 1999) in which the binding motif for the minor spliceosome is reported as AAUUUUUGG. A different variant (A>T) in the same base pair was recently reported in 2 siblings (De Schryver et al. 2016).
The 17G>A mutation is in the Stem II domain, a domain which is implicated in Roifman syndrome exclusively. Since the recent discovery of the genetic cause of this rare syndrome (Merico et al. 2015), it is now possible to obtain a molecular diagnosis. This rare disorder should be considered in any patient with humoral insufficiency, skeletal dysplasia, retinopathy, and developmental delay.
REFERENCES
de Schryver S., Bogaert D., De Baets F., Van Deale S., Schelstraete P., Alizadehfar R., Leroy B.P., Elfride D., Dullaers M., and Haerynck F.2016. Abstracts from the Immunodeficiency Canada—4th SCID Symposium. LymphoSign J.
de Vries P.J., McCartney D.L., McCartney E., Woolf D., and Wozencroft D.2006. The cognitive and behavioural phenotype of Roifman syndrome. J. Intellect. Disabil. Res.50(Pt 9):690–696.
Edery P., Marcaillou C., Sahbatou M., Labalme A., Chastang J., Touraine R., Tubacher E., Senni F., Bober M.B., Nampoothiri S., Jouk P.S., Steichen E., Berland S., Toutain A., Wise C.A., Sanlaville D., Rousseau F., Clerget-Darpoux F., and Leutenegger A.L.2011. Association of TALS developmental disorder with defects in minor splicing component U4atac snRNA. Science.332(6026):240–243.
Fairchild H.R., Fairchild G., Tierney K.M., McCartney D.L., Cross J.J., and de Vries P.J.2011. Parial agenesis of the corpus callosum, hipocampal atrophy and stable intellectual disability associated with Roifman syndrome. Am. J. Med. Genet. Part A.155:2560–2565.
He H., Liyanarachchi S., Akagi K., Nagy R., Li J., Dietrich R.C., Li W., Sebastian N., Wen B., Xin B., Singh J., Yan P., Alder H., Haan E., Wieczorek D., Albrecht B., Puffenberger E., Wang H., Westman J.A., Padgett R.A., Symer D.E., and de la Chapelle A.2011. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD1. Science.332(6026):238–240.
Jafarifar F., Dietrich R.C., Hiznay J.M., and Padgett R.A.2014. Biochemical defects in minor spliceosome function in the developmental disorder MOPD I. RNA.20(7):1078–1089.
Mandel K., Grunebaum E., and Benson L.2001. Noncompaction of the myocardium associated with Roifman syndrome. Cardiol. Young.11:240–243.
Merico D., Roifman M., Braunschweig U., Yuen R.K.C., Alexandrova R., Bates A., Reid B., Nalpathamkalam T., Wang Z., Thiruvahindrapuram B., Gray P., Kakakios A., Peake J., Hogarth S., Manson D., Buncic R., Pereira S.L., Herbrick J.A., Blencowe B.J., Roifman C.M., and Scherer S.W.2015. Comound heterozygous mutations in the noncoding RNU4ATAC cause Roifman syndrome by disrupting minor intron splicing. Nat. Commun.6:8718.
Raker V.A., Hartmuth K., Kastner B., and Lührmann R.1999. Spliceosomal U snRNP core assembly: Sm proteins assemble onto an Sm site RNA nonanucleotide in a specific and thermodynamically stable manner. Mol. Cell. Biol.19(10):6554–6565.
Robertson S.P., Rodda C., and Bankier A.2000. Hypogonadotrophic hypogonadism in Roifman syndrome. Clin. Genet.57:435–438.
Roifman C.M.1997. Immunological aspects of a novel antibody deficiency with normal immunoglobulins, spondyloepiphyseal dysplasia, growth and developmental delay abd retinal dystrophy. Can. J. Allergy Clin. Immunol.2:94–98.
Roifman C.M.1999. Antibody deficiency, growth retardation, spondyloepiphyseal dysplasia and retinal dystrophy: A novel syndrome. Clin. Genet.55:103–109.
Shukla G.C., Cole A.J., Dietrich R.C., and Padgett R.A.2002. Domains of human U4atac snRNA required for U12-dependent splicing in vivo. Nucleic Acids Res.30(21):4650–4657.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 4 • December 2016
Pages: 159 - 163
History
Received: 15 October 2016
Accepted: 23 November 2016
Accepted manuscript online: 23 November 2016
Copyright
© 2016.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Yael DinurSchejter, DanieleMerico, DavidManson, BrendaReid, and LindaVong. 2016. A novel mutation in Roifman syndrome redefines the boundaries of the Sm protein-binding site. LymphoSign Journal.
3(4): 159-163. https://doi.org/10.14785/lymphosign-2016-0015
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
