Whole exome and genome sequencing for Mendelian immune disorders: from molecular diagnostics to new disease variant and gene discovery
Abstract
Whole exome and whole genome sequencing are next generation sequencing (NGS) applications that enable investigation of all coding variants (around 20 000) or all variants (around 4 million) in the human genome. They provide an extremely powerful tool for detecting variants with an established implication in Mendelian disorders as well as for discovering new disease variants and genes. The large number of variants generated requires elaborate databases, prediction models, and integrated workflows to identify which variants are more likely to contribute to disease. We discuss the whole exome and whole genome options, review the sequencing platforms and variant calling pipelines available for different variant types, and devote most of the review to how genetic variants can be annotated and prioritized to identify the ones likely contributing to disorder. The application focus will be Mendelian disorders; disorders caused by rare or common variants with a more complex genetic architecture will only be discussed briefly. For variant annotation and interpretation, we will concentrate on smaller variants (substitutions, insertions, and deletions), only briefly reviewing structural and copy number variation.
Sequencing and variant detection
In this section, we will review how the sequencing process works, what sequencing platforms are available and what analytical methods are typically used to detect genetic variants.
Overview of next generation sequencing of whole exome and genome
Next generation sequencing (NGS) of exomes and genomes typically consists of the following steps (Pabinger et al. 2014). DNA fragments (length 300–500 base pairs (bp)) are processed into a sequencing library, adding adaptor and barcodes to their ends, and then both ends of the fragments are sequenced to produce read pairs, with each read spanning 100–150 bp and the paired reads separated by a gap (Goodwin et al. 2016). Reads are then aligned to the human genome reference sequence by a “read aligner” (e.g., using BWA, the Burrows–Wheeler Aligner (Li and Durbin 2010)). Afterwards, genetic variants (small substitutions, insertions, and deletions) are detected using multi-module tools called “variant callers” (e.g., GATK, the Genome Analysis Toolkit (McKenna et al. 2010)) that receive in input aligned reads and evaluate if the read bases at each reference genome position are supportive of variation, modelling base sequencing quality, alignment specificity, and other quality parameters. Recent-generation variant callers are also able to perform local de-novo sequence assembly, achieving greater sensitivity in detecting longer and more complex variants (e.g., the GATK Haplotype Caller). Specialized variant callers are required for copy number variant (CNV) and other structural variant (such as inversions and translocation) detection (Pabinger et al. 2014). Small substitutions, especially single nucleotide variants (SNVs), are more straightforward to detect; in contrast, insertions, deletions, structural variants, and CNVs are more challenging (Pang et al. 2014). Community efforts have led to the establishment of a variant detection benchmark for small substitutions, insertions, and deletions (Zook et al. 2014), whereas no unified benchmark is available for copy number variants, although several have been proposed (Pang et al. 2014; Mu et al. 2015; Zarrei et al. 2015).
Specialized filtering methods (Jiang et al. 2013; Yuen et al. 2015) or variant callers (e.g., DeNovoGear (Ramu et al. 2013)) are additionally required for detecting if variants arise de-novo in a proband compared to the parental genomes (relevant for trios and other family studies). Finally, variants are annotated for their effect on gene products and allele frequency in established reference datasets; clinical effect classification is fetched from reference databases; additional gene annotations and global probabilistic scores can also be added to facilitate the prioritization of disease causing variants.
Whole exome versus whole genome sequencing
Whole exome differs from whole genome because specific regions, corresponding to coding exons and adjacent genomic intervals, are captured by hybridization probes (or amplified by polymerase chain reaction, depending on the sequencing technology) (Bamshad et al. 2011). Although termed “whole” exome, it is important to note that not all exonic regions are properly captured or amplified because of design and other technical limitations (Bamshad et al. 2011; Jiang et al. 2013). In contrast, whole genome sequencing is able to sequence >96% of the human genome reference sequence (Jiang et al. 2013; Yuen et al. 2015; Stavropoulos et al. 2016). Finally, sequencing depth is more evenly distributed (Jiang et al. 2013). Whole exomes are typically sequenced at average coverage 50–100×, whereas whole genomes are typically sequenced at 30–50× for disease variant identification applications (polymorphism detection when sequencing large sets of individuals can be performed successfully at a much lower depth (1000 Genomes Project Consortium et al. 2015)).
Sequencing platforms
Illumina sequencing platforms currently represent the largest share of the exome and sequencing applications (Goodwin et al. 2016). Whole exome sequencing is typically performed on the HiSeq2500, HiSeq3000, HiSeq4000 instruments; whole genome sequencing can be performed on the same instruments, but is more economic on the newer HiSeqX platform, which offers greater throughput. All instruments of the Illumina HiSeq series perform sequencing by synthesis (SBS) with cyclic reversible termination and 4 colour labels; this approach has an overall accuracy rate of >99.5%, with an overall tendency towards substitution errors and some under-representation of regions with extremely low or extremely high GC content (Goodwin et al. 2016). Exome and whole genome sequencing are typically performed using paired end reads, with each read spanning 100–150 bp and fragments spanning 300–500 bp (Goodwin et al. 2016).
High-quality whole genome sequencing can also be performed using the Complete Genomics platform. Complete Genomics sequencing is based on sequencing by ligation; this approach results in a very high accuracy (∼99.99%), as each base is probed multiple times (Goodwin et al. 2016). Complete Genomics fragments are 500 bp, with 35 bp at both ends covered by shorter reads separated by small gaps and a larger gap in the central portion of the fragment. The Complete Genomics sequencing service includes delivery of called variants, including small substitutions, insertions, deletions, as well as structural and copy number variants. However, the Revolocity sequencing system was intended to compete with the Illumina HiSeq but its launch was suspended in 2016, creating some uncertainty on the future of this sequencing platform (Goodwin et al. 2016).
LifeTech’s AmpliSeq IonProton offers an alternative option for exome and targeted sequencing. This platform relies on SBS with single-nucleotide addition and consequently presents a higher insertion and deletion error rate (Goodwin et al. 2016); if this is not properly addressed by removing lower quality variants, it can lead to many incorrect candidate disease variants with damaging effects. The future of this platform is also less certain, considering the constant improvement and cost reduction of Illumina platforms.
All sequencing platforms described so far are based on short reads; while they currently offer the best value-for-money in terms of exome and whole genome sequencing, they are insufficient for resolving more complex genomic loci with higher repeat content and (or) very high variation between individuals. Single-molecule long-read sequencing, as offered by the Pacific BioSciences RS II system, addresses this problem by offering read lengths of about 20 kb; however the sequencing cost per Gb is much higher compared to the Illumina instruments (20–100× for the PacBio RS II), limiting its applications. This may change in the future as newer system with greater throughput and lower costs are deployed. A cheaper alternative is also offered by synthetic long-read technologies, although long-read reconstruction can be affected by errors (Goodwin et al. 2016).
Other sequencing systems, like SOLiD (colour-space) and 454 have been relegated to a small niche within the industry or recently discontinued and thus they are not described in detail; the BGISEQ-500 system has less information available and thus is not described in detail either (Goodwin et al. 2016).
Variant annotation and prioritization
The large number of variants generated requires elaborate databases, prediction models, and integrated workflows to identify a small number of candidate variants that are more likely to contribute to disease.
Identification of clinically relevant variants is characterized by stricter criteria: it requires very clear and highly impactful gene product effects (e.g., truncating loss-of-function) or functional validation experiments for variants whose effect is more questionable, frequency lower than expected based on disease prevalence, family segregation (ideally from an extended pedigree and (or) multiple independent families) or markedly higher incidence in disease carriers compared to controls, clustering with already established pathogenic mutations, and obviously it is restricted to genes with a well-established implication in Mendelian disease. The ACMG guidelines represent the most broadly adopted human clinical genetics standard for classifying variants as “benign”, “likely benign”, “uncertain significance”, “likely pathogenic”, or “pathogenic” (Richards et al. 2015).
Discovery of novel disease variants within established Mendelian genes follows similar principles, especially as far as frequency filtering, but it is also more open and exploratory in terms of variant effect and impact. Certain effects may be under-reported in clinical databases (e.g., splicing sequence changes besides the highly conserved dinucleotide, transcriptional regulatory changes), and obviously functional validation is typically not already available and may have to be performed for the most promising candidate variants; similar considerations apply to segregation in multiple families. Discovery of variants in novel genes additionally requires prioritization of genes that are likely to contribute to human Mendelian disease based on phenotypic abnormality in model organisms, known functional role, and genetic constraint.
We argue that these different use cases can all be served by bioinformatics workflows prioritizing variants along different “dimensions” (Figure 1): (i) sequencing quality, (ii) allele frequency, (iii) variant effect on gene product, and (iv) gene information. In the presence of trio or other family-based designs, the variant inheritance pattern or de-novo status (v) can also be used for prioritization (Bamshad et al. 2011; Saunders et al. 2012; Jiang et al. 2013; Ngan et al. 2014; Dewey et al. 2015; Merico et al. 2015a, 2015b; Miller et al. 2015; Smedley and Robinson 2015; Yuen et al. 2015; Stavropoulos et al. 2016).
Figure 1:
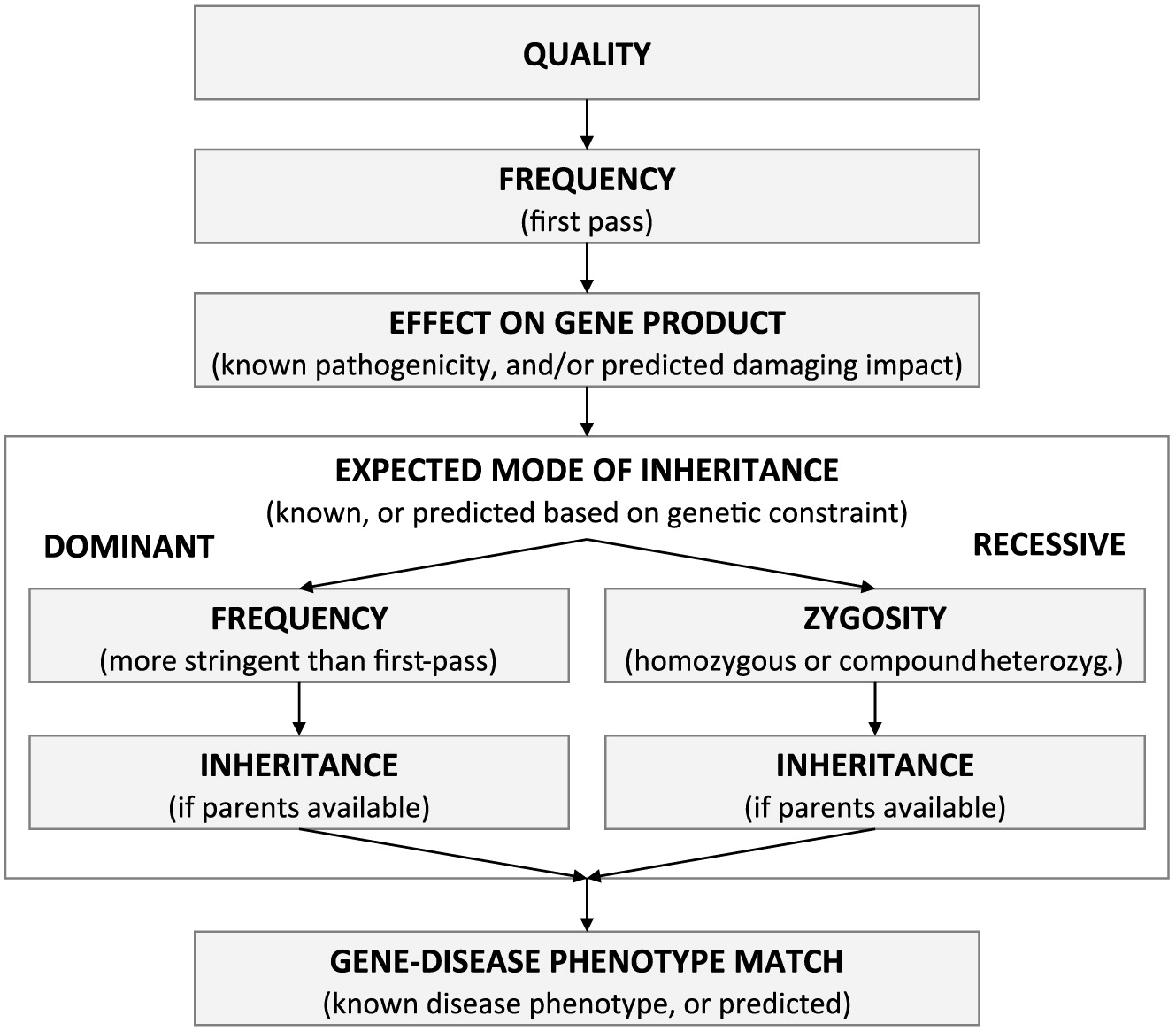
Sequencing quality
For Illumina sequencing analyzed using GATK, the VQSR (Variant Quality Score Recalibration) filter value “PASS” is typically used as the primary quality filter; the VQSR procedure takes advantage of known polymorphisms in the human population to learn what parameter values discriminate true variants from false variants (McKenna et al. 2010). Additional hard filters can be applied to mask regions of the genome that are repeat rich and (or) prone to mapping artifacts (e.g., segmental duplications). Different sets of filters are optimal for Complete Genomics whole genome sequencing (Merico et al. 2015a; Yuen et al. 2015; Stavropoulos et al. 2016). In general, optimal filtering criteria can be established by sequencing the reference sample NA12878 and comparing called variants to the multi-platform benchmark established by the Genome in a Bottle Consortium in collaboration with the National Institute of Standards and Technology (Zook et al. 2014). Additional benchmarks are being established to avoid overfitting on the reference sample (e.g., the PrecisionFDA truth challenge https://precision.fda.gov/challenges/truth).
Allele frequencies
Using a variant filter based on allele frequency in reference databases avoids selecting variants incorrectly predicted to be disruptive for the gene product (Richards et al. 2015). One to five percent is typically used for autosomal recessive genes and <0.1% (or never observed) for dominant genes (Bamshad et al. 2011). The 1000 Genomes project (totalling 2504 individuals in the latest release) is the main resource covering the whole human genome and different continental population (Europeans, Africans, Eastern Asians, Southern Asians, admixed Latin Americans) (1000 Genomes Project Consortium et al. 2015). It is a common practice to filter based on the maximum frequency observed across the 5 continental populations (Bamshad et al. 2011; Lek et al. 2016); while the earlier release of 1000 Genomes presented excessively small continental population sizes for accurate allele frequency estimate, the current size is satisfactory (Brown et al. 2016).
Exome Aggregation Consortium (ExAC) is an additional resource that is restricted to coding regions, but includes many more subjects (60 706 unrelated individuals); caution needs to be exerted when using ExAC frequencies, because ExAC includes disease carrier cohorts, although individuals affected by Mendelian early-onset disorders have been excluded (Lek et al. 2016).
In contrast, it is not advisable to filter variants based on presence or absence in NCBI dbSNP, because dbSNP is a comprehensive variant archive that includes pathogenic variantsand is not restricted to polymorphic variation present in the human population (Bamshad et al. 2011; Johnston and Biesecker 2013).
Finally, certain variants may have gone undetected in reference allele frequencies databases because they are platform-specific artifacts, because they belong to an error-prone or highly variable region that has been masked, or because they are complex and thus their detection and representation are highly dependent on the alignment and variant calling procedure. It is always advisable to include internal platform-matched and analysis pipeline-matched controls to neutralize this type of issue (Bamshad et al. 2011).
Clinical variant classification databases
ClinVar (Landrum et al. 2016) and HMGD (Stenson et al. 2014) are the most popular clinical variant classification databases.
ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) is hosted by the National Center for Biotechnology Information (NCBI) and is freely available. ClinVar currently holds >125 000 classified variants, with almost 4800 genes having variants that affect a single gene (i.e., after removing multi-genic structural variants). The main classification categories of clinical relevance are benign, likely benign, uncertain significance, likely pathogenic, and pathogenic. Clinical variant classification and supporting information are submitted by clinical testing laboratories, research laboratories, and authoritative databases such as OMIM (Online Mendelian Inheritance in Man). Although many submitters follow rigorous guidelines for variant identification (ACMG Guidelines (Richards et al. 2015)), or follow similar guidelines established within their institution (Eggington et al. 2014), conflict in variant classification can arise in the presence of multiple classifications by different submitters.
HGMD is a commercial database (Stenson et al. 2014) with variant entries added by professional curators; it captures variants that are causative or associated to human disease, and tends to favour inclusiveness over stringency in pathogenicity assessment.
In general, clinical classification databases are not perfect: there is a significant degree of disagreement between databases because of different classification standards (Vail et al. 2015). In addition, for a given individual genome, several variants reported as pathogenic can occur in absence of the expected disease, suggesting that they are incorrectly classified or have fairly low penetrance (Xue et al. 2012; Berg et al. 2013; MacArthur et al. 2014; Lek et al. 2016). Obviously, even if a recessive variant is correctly labelled as pathogenic, the zygosity needs to be taken into account (a single heterozygous damaging variant is insufficient to produce disease in an individual in absence of compound heterozygosity). For all these reasons, a variant classified as pathogenic in a database cannot be automatically interpreted as pathogenic when observed in an individual: all lines of evidence (allele frequency, gene product effect, etc.) need to be evaluated (Berg et al. 2013; Richards et al. 2015).
Gene product effect and impact
To understand the effect of genetic variants on downstream molecular and cellular processes, it is important to first determine the variant effect on gene products. While non-genic sequences are important for gene expression regulation, gene products are the main link between the genome and downstream functional processes; effects on regulatory sequences (and how to link them back to the gene product they are presumed to regulate) will be discussed in a dedicated subsection. For clarity, we will use “effect” to refer to the type of gene product alteration, and “impact” to refer to the quantitative extent of the gene product alteration. We will refer to a gene product alteration as “damaging” when its (predicted) impact corresponds to a significant gene product alteration that is typically sufficient to cause disease given the appropriate combination of zygosity and gene mode of inheritance (“pathogenicity”) (MacArthur et al. 2014).
Coding gene effects: overview
Protein-coding genes represent the best functionally characterized subset, and genes implicated in Mendelian disorders are almost all protein coding: protein-coding gene variants represent 99.9% of ClinVar pathogenic or likely pathogenic variants.
Effects on protein-coding genes can be articulated in the following categories: (i) acquisition of a premature stop codon (“stop-gain”), (ii) change in the reading frame (“frameshift”) typically caused by insertions or deletions, (iii) alteration of the highly conserved intronic dinucleotide of a splice site (“core splice site alteration”), (iv) loss of the start codon (“start-loss”), (v) loss of the stop codon (“stop-loss”), (vi) changes of a single amino acid (“missense”), (vii) insertion or deletion or multiple substitutions of amino acids (“in-frame amino acid sequence change”), (viii) changes in the splicing consensus sequence or other sequences regulating splicing (“splicing sequence change”), and (ix) UTR (untranslated sequence) changes with different molecular regulatory mechanisms at 3′ and 5′ UTRs (“UTR change”). It is also common to annotate variants that do not change the amino acid encoding of a codon as “synonymous”; however, these variants may have an effect on splicing, and thus this characterization can be misleading if automatically interpreted as silent or neutral (Richards et al. 2015).
The effect of a variant depends on the gene models and software used (McCarthy et al. 2014; Frankish et al. 2015). NCBI RefSeq, GENCODE Basic, and GENCODE Comprehensive (used by the Ensembl project) are the most commonly used gene models. Gene models from GENCODE Basic and especially from the curated subset of RefSeq (i.e., excluding transcript models, marked as XM_ or XR_) are more conservative, whereas the full RefSeq and especially the GENCODE Comprehensive gene models are more complete, but they also include more transcripts that, even if expressed at sufficiently high levels, can be more functionally questionable (Frankish et al. 2015). It is also important to note that GENCODE Basic does not include non-coding transcripts, whereas the curated subset of RefSeq does. Additionally, GENCODE Comprehensive includes incomplete transcripts, which typically lack a proper start or end and consequently they do not have a proper coding sequence (Frankish et al. 2015).
The terminology used to express the gene product effect also depends on the bioinformatics annotation tool used; the most commonly used are Annovar (Wang et al. 2010), VEP (Variant Effect Predictor) (McLaren et al. 2016), and SnpEff (Cingolani et al. 2012). Sequence Ontology (SO) is a standardized vocabulary for variant effects on gene product (Reese et al. 2010); more systematic adoption of SO terms should lead to uniform effect annotation given the same gene models.
Coding gene effects: truncating loss-of-function variants
Stop-gains and frameshifts are the effects with the strongest impact on the gene product: unless they occur in the terminal part of the coding sequence, they are very likely to trigger nonsense-mediated decay and lead to complete loss of the transcript carrying the variant, resulting in completely abrogated gene product expression for bi-allelic variants. For this reason, these variants referred to as “truncating” or “loss-of-function” (we will refer to them as “truncating loss-of-function” to avoid any ambiguity). Alterations of core splice sites can produce the same outcome by causing exon skipping, skipping of part of the exon, or retention of intronic sequence; alterations of other splicing control sequences can have similar effects, but typically they do not completely abrogate the wild-type transcript and thus need specific predictive models for impact prediction (these will be described in a specific subsection). Start-losses are also often included in the truncating loss-of-function category, although an alternative downstream start site could be utilized.
Impact prediction is typically not necessary for truncating loss-of-function variants (95.6% of ClinVar variants in this category are (likely) pathogenic); however, it is beneficial to consider the percentage of coding sequence truncated and the percentage of transcript isoforms affected.
Coding gene effects: missense and in-frame amino acid changes
Changes of single or multiple amino acids can have very different effects on gene product function depending on the sequence affected and the properties of wild-type and mutant amino acids. At a given site, only a dramatic amino acid change may have a damaging impact (for instance, the ClinVar entry NM_000032.4(ALAS2):c.1676G>C (p.Arg559Pro) is deemed likely pathogenic for hereditary X-linked sideroblastic anemia while NM_000032.4(ALAS2):c.1676G>A (p.Arg559His) is deemed likely benign); in contrast, other sites may be sensitive even to apparently moderate amino changes (e.g., STAMBP K303R (Naseer et al. 2016)). Bioinformatics impact predictors attempt to address this problem and are reviewed in detail in this section. In any case, it is always highly recommendable to manually evaluate additionally available information, such as overlap with annotated protein domains (available from the UCSC Genome Browser via the PFAM and SwissProt tracks), as well as presence of hotspots of pathogenic variants.
Impact predictors are typically available for missense effects, although the same predictive principles could be extended to other amino acid changes. Different predictors can be categorized based on (i) what predictive features are used to predict impact, (ii) whether they require a training set of known damaging or pathogenic variants, and (iii) what such dataset they use. All the most commonly used predictors (such as SIFT (Ng and Henikoff 2001), PolyPhen2 (Adzhubei et al. 2010) and MutationAssessor (Reva et al. 2011)) rely on protein sequence position-specific conservation and amino acid substitution rates based on multiple protein sequence alignments. This is more powerful than genomic DNA conservation or substitution rates due to the degenerate nature of the genetic code and different effects of specific third-base codon nucleotide substitutions. SIFT was the first predictive model using such features, and its simplicity as well as its independence from variants already classified as pathogenic or damaging still make it appealing. MutationAssessor improves the SIFT algorithm by better modelling positions conserved only in protein subfamilies, and similar to SIFT it does not require a training set of already classified variants. In contrast to SIFT and MutationAssessor, PolyPhen2 includes a richer set of predictive features, including not only alignment-derived conservation and substitution rates but also amino acid physicochemical properties and predicted secondary structure; however, that comes at the cost of relying on training data to fit the predictive model, which is (at least partially) undesirable because training data are not completely unbiased. PolyPhen2 predictions are available for two training sets, “HumDiv” and “HumVar”.
Missense impact predictors are far from being perfect (MacArthur et al. 2014). Different types of circularity have been pointed out (Grimm et al. 2015), and the evaluation sets used so far either overlap with the training set of some methods or they are limited to a few genes and types of protein functionality (e.g., enzymatic activity) thus lacking generality. In addition, current generation predictors are incapable of returning more mechanistic details, because systematic data on different types of mechanistic protein structure perturbation are not systematically available yet; therefore, training sets typically consist of pathogenic versus benign or evolutionarily constrained versus evolutionarily diverged variants. In particular, current predictors fail to distinguish between loss-of-function and gain-of-function variants, a particularly vexing issue for autosomal dominant genes that cause Mendelian disorder only upon loss or gain-of-function (e.g., CARD11 gain-of-function (Chan et al. 2013)). Consequently, the use of missense predictors is quite different in a clinical diagnostic or discovery setting; ACMG Guidelines attribute the lowest level of evidence to them and the guidelines require a perfect consensus among different methods (Richards et al. 2015), favouring functional evidence or segregation patterns. In contrast, in discovery settings, it is common to rank candidate missense variants based on the combination of different predictors’ scores.
Beyond the typical approach to missense impact prediction, post-translational modifications (PTMs) such as phosphorylation, ubiquitination, and acetylation are important for protein signalling pathways; identifying variants disrupting them (Reimand et al. 2013; Narayan et al. 2016) represent an area of growing interest, supported by experimental databases (Hornbeck et al. 2012) and bioinformatics predictive models (Wagih et al. 2015).
Coding gene effects: splicing sequence changes
Pre-mRNA splicing is a key cellular process required for gene expression, and alternative splicing regulation enables the expression of a diversified protein repertoire across cell types and environmental conditions (Scotti and Swanson 2016; Sibley et al. 2016). Diverse estimates for the importance of splicing alterations in Mendelian disorders have been reported. Excluding missense variants for simplicity, 8.6%–9.5% of ClinVar pathogenic and likely pathogenic variants are expected to act via splicing alterations of core splice sites or splicing sequence. Of those, the majority (corresponding to 6.8% of ClinVar (likely) pathogenic variants) consist of core splice alterations; in this review, they were grouped with truncating loss-of-function variants because they are typically damaging and do not require impact prediction models (93.7% of ClinVar core splice alterations are pathogenic or likely pathogenic). However, splicing alterations may be under-represented in ClinVar (because of biased focus on coding sequence (Sibley et al. 2016)) and have been estimated to contribute to 16% (Mort et al. 2014) or even more (Soukarieh et al. 2016) of (likely) pathogenic variants in Mendelian disorders, with the difference mostly accounted by unrecognized splice sequence changes. It is also possible that splicing sequence changes may typically have more subtle effects, and thus be more important for disorders with more complex genetic architectures (Merico et al. 2015c; Xiong et al. 2015; Yuen et al. 2016). Finally, while the nervous system is expected to exhibit the largest splicing diversity (Sibley et al. 2016), splicing may be important also for the immune system considering its rich and highly diversified cell type as well as recognition molecule array.
Splicing predictors for splicing sequence alterations can be categorized along 3 axes: (i) whether they are derived from reference data on physiological splicing or directly trained on pre-classified mutation data (pathogenic, benign); (ii) what type of splicing outcome they predict; and (iii) what type of splicing sequence change they are able to model. Models trained on already classified variants typically predict a generic damaging impact without providing mechanistic details on the splicing alteration; other models predict the difference in consensus sequence strength, exon inclusion percentage change, or alternative splice site usage, but they do not predict downstream events such as frameshift, intron retention, or premature stop-gain acquisition, which can result from splicing alteration. Models trained on physiological splicing data are preferable (at least in principle) because they are not prone to overfitting on biases affecting existing labelled mutation data (Xiong et al. 2015). Splicing sequence changes can be categorized as follows: (i) alteration of consensus sequence, important for the constitutive splicing machinery and proximal to the core splice sites; (ii) loss or gain of exonic splicing enhancers and suppressors, often modelled using exhamer-based models; (iii) loss or gain of intronic splicing enhancers and suppressors; (iv) loss or gain of other motifs for splicing factors and RNA-binding proteins; and (v) creation of de-novo core splice sites or strengthening of existing cryptic splice sites.
MaxEntScan (Yeo and Burge 2004) is one of the most popular and effective models for consensus sequence alteration. While it is trained on known and decoy splice site sequence, it performs very well on pathogenic variant classification (Jian et al. 2014). However, it is not able to model the branch site sequence, which is outside of its consensus sequence boundaries.
dbscSNV is another model for consensus sequence alteration. It is trained on mutation data labelled as pathogenic or benign. It uses MaxEntScan and other sequence-based models plus genomic conservation, but it is not able to model the branch site sequence either; it has a performance similar to MaxEntScan alone (Jian et al. 2014).
Several models capture exonic splicing enhancers and suppressors using hexamers (i.e., sequences of 6 nucleotides) (Erkelenz et al. 2014; Rosenberg et al. 2015) and are thus complementary to models targeting consensus sequence. HEXplorer predicts percentage exon inclusion based on hexamer weights learnt by comparing exonic to intronic sequence (Erkelenz et al. 2014). HAL predicts exon inclusion and alternative splice site utilization; it is trained on synthetic sequences and transcriptional patterns measured in a minigene system. As it is trained on synthetic mutation data, it is immune to typical biases that affect other methods trained on pathogenic versus benign variants (Rosenberg et al. 2015).
MutPredSplice predicts the splicing damaging impact of exonic variants and is trained on pre-classified variants; its features consist of scores from consensus sequence models, a mutation-based hexamer model, genomic conservation, and other features based on gene annotation (e.g., distance of the variant from an annotated splice site) (Mort et al. 2014).
SPANR predicts percentage exon inclusion for exonic as well as intronic variants; it is trained on splicing patterns in non-constitutive exons. Its features consist of scores from consensus sequence models, a large compendium of splicing factor and RNA-binding protein motifs, RNA accessibility based on secondary structure, and nucleosome positioning (Xiong et al. 2015).
There are also more complex types of splicing that need specialized predictors and are not reviewed in detail here (Sibley et al. 2016).
Coding gene effects: UTR changes
UTR changes can result in altered gene expression. The 5′ UTR is important for translation regulation (Scheper et al. 2007): it contains the sequence guiding the ribosome assembly on the mature transcript (Kozak sequence for cap complex-dependent transcript recognition, or IRES (internal ribosome entry site(s)) for internal cap-independent recognition) and other regulatory elements; its length, GC content, and presence of secondary structure elements modulate ribosome start site recognition and translation rate; it can also contain decoy start codons that interfere with proper translation (uAUG, upstream AUG, and uORF, upstream open reading frames; the latter comprise a short coding sequence terminating in a stop codon) (Chatterjee and Pal 2009; Barbosa et al. 2013). The 3′ UTR contains the polyadenylation signal important for transcript maturation and can also contain sequences modulating transcript stability recognized by RNA binding proteins or miRNAs (Chatterjee and Pal 2009).
Currently, few Mendelian disorders (such as hereditary thrombocytaemia) have been reported to be caused by UTR single nucleotide substitutions, indels, or exon skipping (ClinVar: 0.2% pathogenic or likely pathogenic variants consisting of UTR changes). There is also a lack of quantitative variant impact prediction methods, perhaps also because of the coding exon bias in exome sequencing capture target design (Scheper et al. 2007; Chatterjee and Pal 2009). Consequently, UTR mutations are often ignored. This may change in the near future, also in relation to the availability of functional genomics dataset profiling UTR regulatory sequences and effects on gene expression (Goodarzi et al. 2012) as well as well-established miRNA target prediction methods (such as TargetScan (Agarwal et al. 2015)). In the interim, in absence of more principled predictors, genomic conservation can be used for UTR change impact (Yuen et al. 2016).
It is possible that a small number of UTR-altering variants have a sufficiently large impact that contributes to Mendelian disorder, whereas the majority may have a smaller effect and may be relevant mainly for more complex disorders (Yuen et al. 2016).
Non-coding gene effects and impact prediction
Non-coding RNA (ncRNA) genes do not code for protein sequence but instead produce a RNA molecule that directly exerts a biochemical activity (structural, catalytic, or binding other nucleic acid molecules and thus modulating processes like transcription and chromatin state regulation). ncRNA genes can range from shorter, more conserved, and better functionally characterized species that have a well-established structural or catalytic role (such as small nuclear RNA, snRNA) or a regulatory role (such as miRNA), to more heterogeneously conserved and characterized lincRNA (long intergenic non-coding RNA, lincRNA) (ENCODE Project Consortium 2012; Palazzo and Lee 2015). Variants impacting the former ncRNA category can be efficiently scored using genomic conservation, although specialized models may be beneficial for miRNA; in contrast, for the latter category impact prediction is more challenging and conservation-based criteria may only be partially useful (Chodroff et al. 2010; Derrien et al. 2012).
In general, ncRNA variants causing Mendelian disorders are not many (Quek et al. 2015), and variant effect evaluation may become more efficient only as more sophisticated impact prediction models are developed and more information is accumulated at the gene level. It is also possible that the majority of miRNA and lincRNA variants cause molecular process alterations insufficient for Mendelian disorder causation, but sufficient for pathogenicity contribution in cancer and other complex disorders, whereas only a few highly conserved ncRNA that have a key role in biological processes can effectively contribute to Mendelian disorders (e.g., the small nuclear RNA gene RNU4ATAC (Edery et al. 2011; He et al. 2011)).
Gene transcriptional regulation effect and impact prediction
Transcriptional regulation relies on several intertwined molecular layers: chromatin state and DNA methylation control DNA accessibility to transcription factors and the transcriptional machinery, with different functional elements (e.g., active promoters, enhancers, insulators) characterized by different histone mark combinations; in turn, specific transcription factors can initiate changes in the local chromatin and DNA methylation state. The completion of large-scale projects such as ENCODE and Roadmap Epigenomics (ENCODE Project Consortium 2012; Kundaje et al. 2015) has resulted in a wealth of data for different cell types, profiling binding-accessible chromatin sites (using DNase-seq or ATAC-seq), histone marks, and transcription factor binding (using ChIP-seq). Chromatin states are typically inferred using histone marks and other available data (such as methylation and binding of specific factors) (Ernst and Kellis 2012; Hoffman et al. 2012).
Certain predictive models focus on transcription factor binding affinity changes induced by sequence changes; position-specific weight matrices have been broadly used to model transcription factor sequence specificity (Wasserman and Sandelin 2004), but recently more powerful models based on deep learning have been introduced (e.g., DeepBind, (Alipanahi et al. 2015)). Additional information, such as occurrence of the predicted binding event in other species or genomic sequence conservation can be used to refine predictions (Wenger et al. 2013). Cell-specific inferred chromatin states are also useful to put these predictions in context.
Other predictive methods are trained to directly learn the sequence specificity of selected functional regions, such as DNaseI hypersenstivity sites (expected to correspond to open chromatin protein binding sites) or enhancers (Lee et al. 2015; Zhou and Troyanskaya 2015; Kelley et al. 2016).
Mapping variants to target genes requires more effort compared to other impact predictors. Since chromatin looping can establish physical proximity to target genes for distal regulatory elements, experimentally determined (Denker and de Laat 2016) and predicted chromatin contact maps (Whalen et al. 2016; Zhu et al. 2016) are important.
While a limited number of variants have been implicated in Mendelian disorders so far (Deplancke et al. 2016), this type of variants may have a significant and under-recognized role in Mendelian disorders, perhaps especially in compound heterozygosity with a strong-effect variant (Zhang and Lupski 2015). Despite the increasing availability of predictive models, there are persisting challenges to overcome: (i) experimental benchmarks critical for predictor evaluation are still being defined (e.g., (Maurano et al. 2015)), (ii) the presence of complex interactive effects between different transcription factors prevents a simple reductionist approach to the prediction problem, and (iii) the redundancy of transcriptional regulation prevents the loss of a specific transcription factor binding site may be insufficient to produce abnormal phenotype (Deplancke et al. 2016). Structural variants deleting regulatory elements have also been reported as causing Mendelian disorders (Weedon et al. 2014), and their impact may be easier to interpret, especially when deleting larger portions of the genome, as long as transcriptional regulatory sequence alteration can be correctly mapped to its target gene(s) (Zhang and Lupski 2015).
Genomic conservation measures
The availability of reference sequences for many other mammalian and vertebrate species enables using their multiple sequence alignments for deriving genomic conservation scores. While these scores are overall highly correlated with the presence of coding exonic sequence, they can help discriminate more diverged genes and identify elements outside of coding exons expected to have conserved function (e.g., UTRs, intronic or intergenic regulatory elements, ncRNA genes). The most popular resources are PhyloP (LRT test) (Pollard et al. 2010), PhastCons (Siepel et al. 2005), and GERP++ (Davydov et al. 2010). PhyloP (LRT test) provides a position-specific conservation score, PhastCons provide a regional conservation score used to identify conserved elements, and GERP++ provides both a positional and regional conservation score. In particular, GERP++ conserved elements have been demonstrated to be less fragmented than PhastCons (Davydov et al. 2010). In addition, currently available GERP++ is based only on mammalian genomes, whereas PhyloP (LRT test) and PhastCons are available for primate, mammalian, and vertebrate genomes. Conservation based on mammalian genomes is overall less statistically powerful but is able to identify mammalian-specific elements.
Meta-predictors
Meta-predictors aim to optimally combine different predictors or to train a more comprehensive model, including features used by other predictors as well as impact scores returned by other predictors (e.g., CADD (Kircher et al. 2014), Eigen (Ionita-Laza et al. 2016)). While valuable (especially for less sophisticated users or for statistical methods requiring a single impact score), meta-predictors also present the following issues: (i) challenges in the definition of impact benchmarks prevent from definitively establishing if the meta-predictor is better than each individual specialized predictors (Grimm et al. 2015) and (ii) effects that have stronger impact than others may dominate the predictive performance and make the predictive model less sensitive to more subtle changes.
Gene information: disease phenotype, mode of inheritance, genetic constraint, and prioritization of new disease genes
So far we have reviewed in high detail variant effects on gene products and models predicting their impact. While a variant may be very rare and have a highly damaging impact on the gene product, the corresponding gene may be dispensable, thus resulting in no (or minor) phenotypic abnormality, or the variant may be heterozygous and the gene autosomal recessive, resulting in no phenotype in absence of other damaging or at least hypofunctional variants. Therefore, it is important to additionally rank variants based on the likelihood that a gene alteration will produce the disease phenotype of interest, and to also consider if the variant zygosity and the gene’s known mode of inheritance can cause disease based on simple Mendelian genetic rules. Quantitative models of genetic constraint based on genetic variation in the human population can be used to independently validate the known mode of inheritance and also prioritize new disease genes. Prioritization of new disease genes additionally benefits from known abnormal phenotypes in model organism (caused by knock outs or other engineered genetic constructs), annotated gene function, and experimentally determined gene product interaction networks.
Gene information: disease phenotype
In a clinical diagnostic setting, only variants on established Mendelian disease genes are considered. In a discovery setting, it is reasonable to first consider variants that are likely to disrupt established Mendelian disease genes. In both cases, it is desirable to rank genes by how well their known phenotypic spectrum matches with the patient’s disease.
The Human Phenotype Ontology (HPO) (Kohler et al. 2014) is the main resource for modelling genes’ known phenotypic spectrum in Mendelian disease (Smedley and Robinson 2015). It consists of a structured controlled vocabulary with terms connected by formal relations; the ontology is composed of 3 independent sub-ontologies that capture the mode of inheritance, the onset and clinical course, and phenotypic abnormalities. Gene-HPO term annotations are derived from OMIM, Orphanet, and DECIPHER using automated text mining procedures integrated by human expert curation. Since HPO terms capturing phenotypic abnormalities decompose disease into phenotypic elements at different granularity levels (e.g., “Immunodeficiency” is a subclass of the broader class “Abnormality of the immune system”), they can be used by algorithms computing the degree of phenotypic match between the gene and the patient’s presentation (Kohler et al. 2009). Other available algorithms for phenotypic matching take into account additional evidence such as model organism abnormal phenotypes for orthologous genes, allele frequency, variant effect and impact, gene function and interaction networks, family segregation and mode of inheritance (Phevor (Singleton et al. 2014), Exomiser (Smedley et al. 2015), PhenIX (Zemojtel et al. 2014), and comparative performance reviewed by Smedley and Robinson (2015)). These algorithms are more appropriate for discovery rather than clinical diagnostic settings; in addition, while they offer a final aggregate rank, they may give less control to the user in fine tuning different lines of evidence or modeling additional effects and impact predictors. More details are presented in the “Gene annotation: prioritizing new disease genes” subsection.
Gene mode of inheritance and variant zygosity required for disease causation
The concept of dominance is central to Mendelian genetics. Disease causation for genes with a reported autosomal recessive mode of inheritance requires a damaging homozygous variant or compound heterozygosity with (at least) 2 damaging variants. X-linked genes are typically not dominant, and hemizygous damaging variation in males will cause disease, while heterozygous females will be normal or present significantly attenuated disease phenotype (with the exception of skewed X inactivation). While disease for autosomal recessive genes and X-linked genes is typically caused by loss-of-function variation, the landscape is more varied for dominant genes: heterozygous loss-of-function (i.e., haploinsufficient), gain-of-function, and more rarely dominant-negative or toxic-gain-of-function can all be causative of disease, with specific genes or sometimes specific domains within genes following the same mechanism. Mode of inheritance is available from CGD (Clinical Genomic Database) (Solomon et al. 2013) and HPO. However, the detailed indication of mechanism (loss-of-function, gain-of-function, and more rarely dominant-negative or toxic-gain-of-function) is not available and needs to be mined from the literature or textual summaries of the literature available in authoritative databases such as OMIM.
Gene information: modelling genetic constraint
Measures of genetic constraint can be used to select genes that are under negative selection for variation and thus more likely to produce a phenotype if their gene product is altered. Their use is particularly helpful for discovering new disease genes, although they can also be used to flag Mendelian genes with limited constrained (thus more likely to be prone to predicted damaging yet benign variation) or further refine dominant genes based on sensitivity to truncating (i.e., haploinsufficient) as opposed to missense variants. For new disease gene discovery, constraint metrics are complementary to phenotypic information in model organisms and functional information from Gene Ontology (Gene Ontology Consortium et al. 2013), pathways, and networks. Haploinsufficiency prediction is a particularly interesting problem because model organism phenotypes are more often available only for homozygous loss-of-function constructs, and established dominant Mendelian genes are not characterized as haploinsufficient or gain-of-function by available resources (CGD, HPO).
The haploinsufficiency prediction model proposed by Huang et al. (2010) uses genomic characteristics and gene network interactions as predictive features; known haploinsufficient genes mined from the literature constitute the positive training set, whereas genes with common copy number losses constitute the negative training set. Most of the predictive performance is driven by network proximity to known haploinsufficient genes, thus questioning the validity of the model, which may be highly biased towards interacting clusters of known haploinsufficient genes (Steinberg et al. 2015). A better alternative is offered by the ExAC pLI (Lek et al. 2016), which models the probability of a gene being intolerant to truncating loss-of-function variation (i.e., haploinsufficient); the pLI estimate is derived from the difference between the expected and observed number of truncating loss-of-function variants in ExAC, where the expected number is based on a background mutational model including the variants’ trinucleotide context and corrected for covariates like exome sequencing depth (Samocha et al. 2014; Lek et al. 2016).
The genic intolerance (GI) score (Petrovski et al. 2013) models the constraint of a gene to coding sequence alterations, without distinguishing between recessive and dominant genes or between different effects (i.e., truncating loss-of-function versus more localized amino acid changes). The GI is derived from regression residuals obtained by comparing the gene-wise number of common coding sequence altering variants to the gene-wise total number of coding variants from the NHLBI-ESP6500 exome dataset (Tennessen et al. 2012); this assumes that synonymous variants are never damaging. The ExAC missense constraint z-score probably represents a better alternative, considering ExAC has about 10× more exomes than NHLBI-ESP6500, and the missense constraint score follows a dominant model. Similar to pLI, this score is derived from the difference between the expected and observed number of missense variants in ExAC; however, compared to pLI it is more correlated to gene length. While no constraint score is available for gain-of-function dominant variation, genes with high ExAC missense constraint and low truncating loss-of-function constraint (pLI) can be presumed to mainly cause disease via gain-of-function missense variation.
Other measures of constraint have also been proposed but their use has been more limited or specific to certain variant types (Uddin et al. 2014; Telenti et al. 2016).
Gene information: prioritizing new disease genes
Genes that lack a disease phenotype in humans may have orthologs investigated in model organisms using genetic constructs like homozygous knock-out. One of the most valuable resources is phenotypic abnormality annotations provided by MGI (Mouse Genome Informatics) (Eppig et al. 2015), based on the Mammalian Phenotype Ontology (Smith and Eppig 2012). Since mouse genetic constructs are most often available as homozygous, this resource is particularly valuable for the discovery of novel recessive and X-linked genes.
In absence of direct phenotypic information, additional functional genomics resources can be leveraged to prioritize genes: (i) functional gene-sets, (ii) pathways, (iii) gene interaction networks, and (iv) genetic constraint scores. Gene Ontology (Gene Ontology Consortium et al. 2013) provides a controlled vocabulary for functional annotations, and corresponding gene annotations are available from human gene databases such NCBI Entrez Gene; Gene Ontology annotations can be mined for immune-related functions such as “pre-B cell differentiation” (GO:0002329). Pathway databases provide more structured information than Gene Ontology: pathways consist of metabolic, signalling, or other regulatory molecular processes represented using directed causal interactions among gene products and small molecules (Cary et al. 2005). For instance, the pathway database KEGG (Kyoto Encyclopedia of Genes and Genomes) (Kanehisa et al. 2016) has 16 immune pathways, such as the B-cell and T-cell receptor signalling pathways (hsa04662, hsa04660). Other commonly used pathways databases that include immune pathways are Reactome (Fabregat et al. 2016), WikiPathways (Kutmon et al. 2016) and NetPath (Kandasamy et al. 2010). Pathways are particularly useful for highly focused searches in presence of a strong hypothesis about what metabolic, signalling, or regulatory process is presumed to be altered in the patient. In addition, gene interaction networks (protein–protein physical interaction networks, co-expression, etc.) can be leveraged to expand a set of input genes into a larger set of functionally related genes; when this approach is applied to predict new disease genes from known disease genes, it follows the principle of “guilt by association” (Tranchevent et al. 2011; Wang et al. 2011; Smedley et al. 2014). While gene interaction networks do not present the high level of curation and detail as pathways, they cover many more genes. Manual searches can be carried out using visualization tools like GeneMANIA (Zuberi et al. 2013). In addition, some of the previously reviewed prioritization models utilizing phenotypic match plus additional evidence take advantage of Gene Ontology functional annotations and (or) network interactions (Phevor (Singleton et al. 2014), Exomiser (Smedley and Robinson 2015)).
Study design and diagnostic yield
Study design considerations
For disorders prevalently caused by dominant de-novo variants (mainly observed for neurodevelopmental disorders like intellectual disability (Veltman and Brunner 2012)) the trio design (i.e., parents and affected proband) is the most appropriate (Bamshad et al. 2011; MacArthur et al. 2014). For larger cohorts, statistical modelling capturing gene mutability (Samocha et al. 2014) can be leveraged to identify genes with a significant de-novo mutation load (MacArthur et al. 2014; Deciphering Developmental Disorders Study 2015; Ware et al. 2015).
For dominant inherited disorders, large multi-generation families are the most suitable for variant identification. Genotyping some family members on linkage array (Botstein and Risch 2003) instead of using whole exome or whole genome for all members is cost effective (MacArthur et al. 2014); similarly, it is most beneficial to sample most-distally related individuals within the family (Bamshad et al. 2011).
Autosomal recessive disorders are usually easier to solve (Bamshad et al. 2011); it is important to distinguish between presence and absence of consanguinity. In the presence of consanguinity, the variant is likely to be homozygous and to be embedded within a homozygosity stretch; minimally, one affected individual should be sequenced. Adding the parents, an unaffected sibling, and another affected sibling helps reduce the number of candidate variants and the amount of follow-up work. Finally, genotyping some family members on linkage array instead of using whole exome or whole genome is cost effective. In the absence of consanguinity, the variant can be homozygous or compound heterozygous, and it is recommended to sequence the parents and one affected or unaffected sibling in addition to the affected individual. In both circumstances, sequencing multiple unrelated families strengthens the confidence in the finding, especially for novel Mendelian genes.
For X-linked disorders, sequencing the affected male individual is typically sufficient.
Diagnostic yield considerations
The average diagnostic yield for whole exome sequencing in cohorts of patients referred for clinical sequencing is estimated at 25% (Yang et al. 2014); this captures truncating loss-of-function and amino acid changes, whereas contribution of splicing sequence and UTR changes may be under-estimated. Additionally evaluating copy number variants increases diagnostic yield from 25% to 35% for cohorts with congenital abnormalities and (or) developmental delay, and whole genome sequencing can lead to reliable detection of pathogenic copy number changes (Stavropoulos et al. 2016).
The contribution of UTR, splicing sequence, and transcriptional regulatory sequence changes has been estimated for complex disorders in preliminary studies (Yuen et al. 2016), but there is no robust estimate for Mendelian disorders. The contribution of structural variants to diagnostic yield is still being assessed (Noll et al. 2016).
Conclusions and future directions
Challenges
Coding variant effects are biologically well understood, there is a wealth of models for impact predictions and guidelines for clinical variant classification. Nonetheless, amino acid change impact prediction is not a solved problem (MacArthur et al. 2014; Grimm et al. 2015); distinction between loss-of-function and gain-of-function is a particularly important unsolved problem. Databases with clinically classified variants contain errors (MacArthur et al. 2014). Finally, while there is a consensus that an integrated probabilistic model should be used for clinical and discovery variant evaluation, no such consensus model has been developed (MacArthur et al. 2014).
Whole genome sequencing poses additional challenges. Impact predictors are available for splicing sequence changes and transcriptional regulation changes, but the lack of large and well-established evaluation benchmarks and their underrepresentation in clinical classification databases hamper performance assessment (Smedley et al. 2016); in addition, more advanced predictors than currently available may be required to achieve optimal performance. Comprehensive impact predictors are not available for UTR changes or long non-coding RNA yet. For these 4 effect categories, it is difficult to anticipate how many variants may have a strong enough impact to contribute to Mendelian disorder causation, as opposed to disorders with more complex architecture.
Opportunities: using RNA-seq and epigenetic profiling to facilitate diagnostics and discovery
RNA-seq can be leveraged to aid the detection of splicing-altering variants: in the presence of RNA-seq from control individuals and patients for a disease-relevant tissue, splicing junction detection can reveal junctions present or absent in 1 or more patients but not in the controls (Cummings et al. 2016). This approach can also help detect variants missed by exome capture or suggest the presence of structural variation (Cummings et al. 2016).
In addition, variants damaging core splicing machinery can lead to widespread splicing alteration that can be readily detected by RNA-seq (Argente et al. 2014; Merico et al. 2015a). Following a similar strategy, genome-wide profiling of epigenetic marks by ChIP-seq or methylation arrays may help revealing widespread alterations caused by genetic variants disrupting master regulators of DNA methylation and chromatin modification (Yuen et al. 2016). These approaches can be particularly powerful when the impact of genetic variants is more difficult to predict.
Variants at the interface between Mendelian and more complex genetic architectures
Low-penetrance or variable-expressivity dominant Mendelian variants may benefit from analytical strategies developed for complex disorders. Burden tests can reveal additive effects at the pathway level (Bansal et al. 2010). Polygenic risk score imparted by common variants, which can be calculated in presence of GWAS results on large cohorts, (International Schizophrenia Consortium et al. 2009), can lower the risk threshold for variants with lower penetrance (Merico et al. 2015c).
Acknowledgements
Daniele Merico is a full-time employee of Deep Genomics Inc. and has a scientific affiliation to The Centre for Applied Genomics (TCAG) at The Hospital for Sick Children. Special thanks to Prof. Chaim M. Roifman, Prof. Stephen W. Scherer, Dr. Christian R. Marshall, Dr. Ryan K.C. Yuen, and other TCAG team members for highly valuable scientific discussions on Mendelian and complex disorder genetics research. Special thanks also to Prof. Brendan J. Frey, Dr. Babak Alipanahi, Dr. Andrew Delong, Dr. Jinkuk Kim, Dr. Mark Sun, Omar Wagih, Michael Wainberg, Dr. Hui Yuan Xiong, and other Deep Genomics team members for highly valuable scientific discussions on quantitative modelling of splicing and non-coding variant effects.
REFERENCES
Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., and Sunyaev S.R.2010. A method and server for predicting damaging missense mutations. Nat. Methods.7(4):248–249.
Agarwal V., Bell G.W., Nam J.W., and Bartel D.P.2015. Predicting effective microRNA target sites in mammalian mRNAs. Elife.4.
Alipanahi B., Delong A., Weirauch M.T., and Frey B.J.2015. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol.33(8):831–838.
Argente J., Flores R., Gutierrez-Arumi A., Verma B., Martos-Moreno G.A., Cusco I., Oghabian A., Chowen J.A., Frilander M.J., and Perez-Jurado L.A.2014. Defective minor spliceosome mRNA processing results in isolated familial growth hormone deficiency. EMBO Mol. Med.6(3):299–306.
Baker M.2011. Sorting out sequencing data. Nat. Methods.8(10):799–803.
Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., and Shendure J.2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet.12(11):745–755.
Bansal V., Libiger O., Torkamani A., and Schork N.J.2010. Statistical analysis strategies for association studies involving rare variants. Nat. Rev. Genet.11(11):773–785.
Barbosa C., Peixeiro I., and Romao L.2013. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet.9(8):e1003529.
Berg J.S., Adams M., Nassar N., Bizon C., Lee K., Schmitt C.P., Wilhelmsen K.C., and Evans J.P.2013. An informatics approach to analyzing the incidentalome. Genet. Med.15(1):36–44.
Botstein D. and Risch N.2003. Discovering genotypes underlying human phenotypes: Past successes for Mendelian disease, future approaches for complex disease. Nat. Genet.33(Suppl):228–237.
Brown R., Lee H., Eskin A., Kichaev G., Lohmueller K.E., Reversade B., Nelson S.F., and Pasaniuc B.2016. Leveraging ancestry to improve causal variant identification in exome sequencing for monogenic disorders. Eur. J. Hum. Genet.24(1):113–119.
Cary M.P., Bader G.D., and Sander C.2005. Pathway information for systems biology. FEBS Lett.579(8):1815–1820.
Chan W., Schaffer T.B., and Pomerantz J.L.2013. A quantitative signaling screen identifies CARD11 mutations in the CARD and LATCH domains that induce Bcl10 ubiquitination and human lymphoma cell survival. Mol. Cell. Biol.33(2):429–443.
Chatterjee S. and Pal J.K.2009. Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol. Cell.101(5):251–262.
Chodroff R.A., Goodstadt L., Sirey T.M., Oliver P.L., Davies K.E., Green E.D., Molnar Z., and Ponting C.P.2010. Long noncoding RNA genes: Conservation of sequence and brain expression among diverse amniotes. Genome Biol.11(7):R72.
Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., and Ruden D.M.2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin).6(2):80–92.
Cummings B.B., Marshall J.L., Tukiainen T., Lek M., Donkervoort S., Foley A.R., Bolduc V., Waddell L., Sandaradura S., O’Grady G.L., Estrella E., Reddy H.M., Zhao F., Weisburd B., Karczewski K., O’Donnell-Luria A., Birnbaum D., Sarkozy A., Hu Y., Gonorazky H., Claeys K., Joshi H., Bournazos A., Oates E., Ghaoui R., Davis M., Laing N.G., Topf A., Consortium G., Beggs A., Kang P.B., North K.N., Straub V., Dowling J., Muntoni F., Clarke N.F., Cooper S.T., Bonnemann C.G., and MacArthur D.G.2016. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing.
Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., and Batzoglou S.2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol.6(12):e1001025.
Deciphering Developmental Disorders Study. 2015. Large-scale discovery of novel genetic causes of developmental disorders. Nature.519(7542):223–228.
Denker A. and de Laat W.2016. The second decade of 3C technologies: Detailed insights into nuclear organization. Genes Dev.30(12):1357–1382.
Deplancke B., Alpern D., and Gardeux V.2016. The genetics of transcription factor DNA binding variation. Cell.166(3):538–554.
Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G., Lagarde J., Veeravalli L., Ruan X., Ruan Y., Lassmann T., Carninci P., Brown J.B., Lipovich L., Gonzalez J.M., Thomas M., Davis C.A., Shiekhattar R., Gingeras T.R., Hubbard T.J., Notredame C., Harrow J., and Guigo R.2012. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res.22(9):1775–1789.
Dewey F.E., Grove M.E., Priest J.R., Waggott D., Batra P., Miller C.L., Wheeler M., Zia A., Pan C., Karzcewski K.J., Miyake C., Whirl-Carrillo M., Klein T.E., Datta S., Altman R.B., Snyder M., Quertermous T., and Ashley E.A.2015. Sequence to medical phenotypes: A framework for interpretation of human whole genome DNA sequence data. PLoS Genet.11(10):e1005496.
Edery P., Marcaillou C., Sahbatou M., Labalme A., Chastang J., Touraine R., Tubacher E., Senni F., Bober M.B., Nampoothiri S., Jouk P.S., Steichen E., Berland S., Toutain A., Wise C.A., Sanlaville D., Rousseau F., Clerget-Darpoux F., and Leutenegger A.L.2011. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science.332(6026):240–243.
Eggington J.M., Bowles K.R., Moyes K., Manley S., Esterling L., Sizemore S., Rosenthal E., Theisen A., Saam J., Arnell C., Pruss D., Bennett J., Burbidge L.A., Roa B., and Wenstrup R.J.2014. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin. Genet.86(3):229–237.
ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature.489(7414):57–74.
Eppig J.T., Blake J.A., Bult C.J., Kadin J.A., and Richardson J.E., and Mouse Genome Database Group. 2015. The Mouse Genome Database (MGD): Facilitating mouse as a model for human biology and disease. Nucleic Acids Res.43(Database issue):D726–D736.
Erkelenz S., Theiss S., Otte M., Widera M., Peter J.O., and Schaal H.2014. Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res.42(16):10681–10697.
Ernst J. and Kellis M.2012. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods.9(3):215–216.
Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S., Matthews L., May B., Milacic M., Rothfels K., Shamovsky V., Webber M., Weiser J., Williams M., Wu G., Stein L., Hermjakob H., and D’Eustachio P.2016. The Reactome pathway Knowledgebase. Nucleic Acids Res.44(D1):D481–D487.
Frankish A., Uszczynska B., Ritchie G.R., Gonzalez J.M., Pervouchine D., Petryszak R., Mudge J.M., Fonseca N., Brazma A., Guigo R., and Harrow J.2015. Comparison of GENCODE and RefSeq gene annotation and the impact of reference geneset on variant effect prediction. BMC Genomics.16(Suppl 8):S2.
Gene Ontology Consortium, Blake J.A., Dolan M., Drabkin H., Hill D.P., Li N., Sitnikov D., Bridges S., Burgess S., Buza T., McCarthy F., Peddinti D., Pillai L., Carbon S., Dietze H., Ireland A., Lewis S.E., Mungall C.J., Gaudet P., Chrisholm R.L., Fey P., Kibbe W.A., Basu S., Siegele D.A., McIntosh B.K., Renfro D.P., Zweifel A.E., Hu J.C., Brown N.H., Tweedie S., Alam-Faruque Y., Apweiler R., Auchinchloss A., Axelsen K., Bely B., Blatter M., Bonilla C., Bouguerleret L., Boutet E., Breuza L., Bridge A., Chan W.M., Chavali G., Coudert E., Dimmer E., Estreicher A., Famiglietti L., Feuermann M., Gos A., Gruaz-Gumowski N., Hieta R., Hinz C., Hulo C., Huntley R., James J., Jungo F., Keller G., Laiho K., Legge D., Lemercier P., Lieberherr D., Magrane M., Martin M.J., Masson P., Mutowo-Muellenet P., O’Donovan C., Pedruzzi I., Pichler K., Poggioli D., Porras Millan P., Poux S., Rivoire C., Roechert B., Sawford T., Schneider M., Stutz A., Sundaram S., Tognolli M., Xenarios I., Foulgar R., Lomax J., Roncaglia P., Khodiyar V.K., Lovering R.C., Talmud P.J., Chibucos M., Giglio M.G., Chang H., Hunter S., McAnulla C., Mitchell A., Sangrador A., Stephan R., Harris M.A., Oliver S.G., Rutherford K., Wood V., Bahler J., Lock A., Kersey P.J., McDowall D.M., Staines D.M., Dwinell M., Shimoyama M., Laulederkind S., Hayman T., Wang S., Petri V., Lowry T., D’Eustachio P., Matthews L., Balakrishnan R., Binkley G., Cherry J.M., Costanzo M.C., Dwight S.S., Engel S.R., Fisk D.G., Hitz B.C., Hong E.L., Karra K., Miyasato S.R., Nash R.S., Park J., Skrzypek M.S., Weng S., Wong E.D., Berardini T.Z., Huala E., Mi H., Thomas P.D., Chan J., Kishore R., Sternberg P., Van Auken K., Howe D., and Westerfield M.2013. Gene Ontology annotations and resources. Nucleic Acids Res.41(Database issue):D530–D535.
1000 Genomes Project Consortium, Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., and Abecasis G.R.2015. A global reference for human genetic variation. Nature.526(7571):68–74.
Goodarzi H., Najafabadi H.S., Oikonomou P., Greco T.M., Fish L., Salavati R., Cristea I.M., and Tavazoie S.2012. Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature.485(7397):264–268.
Goodwin S., McPherson J.D., and McCombie W.R.2016. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet.17(6):333–351.
Grimm D.G., Azencott C.A., Aicheler F., Gieraths U., MacArthur D.G., Samocha K.E., Cooper D.N., Stenson P.D., Daly M.J., Smoller J.W., Duncan L.E., and Borgwardt K.M.2015. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat.36(5):513–523.
He H., Liyanarachchi S., Akagi K., Nagy R., Li J., Dietrich R.C., Li W., Sebastian N., Wen B., Xin B., Singh J., Yan P., Alder H., Haan E., Wieczorek D., Albrecht B., Puffenberger E., Wang H., Westman J.A., Padgett R.A., Symer D.E., and de la Chapelle A.2011. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science.332(6026):238–240.
Hoffman M.M., Buske O.J., Wang J., Weng Z., Bilmes J.A., and Noble W.S.2012. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods.9(5):473–476.
Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., and Sullivan M.2012. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res.40(Database issue):D261–D270.
Huang N., Lee I., Marcotte E.M., and Hurles M.E.2010. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet.6(10):e1001154.
International Schizophrenia Consortium, Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O’Donovan M.C., Sullivan P.F., and Sklar P.2009. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature.460(7256):748–752.
Ionita-Laza I., McCallum K., Xu B., and Buxbaum J.D.2016. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet.48(2):214–220.
Jian X., Boerwinkle E., and Liu X.2014. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res.42(22):13534–13544.
Jiang Y.H., Yuen R.K., Jin X., Wang M., Chen N., Wu X., Ju J., Mei J., Shi Y., He M., Wang G., Liang J., Wang Z., Cao D., Carter M.T., Chrysler C., Drmic I.E., Howe J.L., Lau L., Marshall C.R., Merico D., Nalpathamkalam T., Thiruvahindrapuram B., Thompson A., Uddin M., Walker S., Luo J., Anagnostou E., Zwaigenbaum L., Ring R.H., Wang J., Lajonchere C., Wang J., Shih A., Szatmari P., Yang H., Dawson G., Li Y., and Scherer S.W.2013. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet.93(2):249–263.
Johnston J.J. and Biesecker L.G.2013. Databases of genomic variation and phenotypes: Existing resources and future needs. Hum. Mol. Genet.22(R1):R27–R31.
Kandasamy K., Mohan S.S., Raju R., Keerthikumar S., Kumar G.S., Venugopal A.K., Telikicherla D., Navarro J.D., Mathivanan S., Pecquet C., Gollapudi S.K., Tattikota S.G., Mohan S., Padhukasahasram H., Subbannayya Y., Goel R., Jacob H.K., Zhong J., Sekhar R., Nanjappa V., Balakrishnan L., Subbaiah R., Ramachandra Y.L., Rahiman B.A., Prasad T.S., Lin J.X., Houtman J.C., Desiderio S., Renauld J.C., Constantinescu S.N., Ohara O., Hirano T., Kubo M., Singh S., Khatri P., Draghici S., Bader G.D., Sander C., Leonard W.J., and Pandey A.2010. NetPath: A public resource of curated signal transduction pathways. Genome Biol.11(1):R3.
Kanehisa M., Sato Y., Kawashima M., Furumichi M., and Tanabe M.2016. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res.44(D1):D457–D462.
Kelley D.R., Snoek J., and Rinn J.L.2016. Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res.26(7):990–999.
Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., and Shendure J.2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet.46(3):310–315.
Kohler S., Doelken S.C., Mungall C.J., Bauer S., Firth H.V., Bailleul-Forestier I., Black G.C., Brown D.L., Brudno M., Campbell J., FitzPatrick D.R., Eppig J.T., Jackson A.P., Freson K., Girdea M., Helbig I., Hurst J.A., Jahn J., Jackson L.G., Kelly A.M., Ledbetter D.H., Mansour S., Martin C.L., Moss C., Mumford A., Ouwehand W.H., Park S.M., Riggs E.R., Scott R.H., Sisodiya S., Van Vooren S., Wapner R.J., Wilkie A.O., Wright C.F., Vulto-van Silfhout A.T., de Leeuw N., de Vries B.B., Washingthon N.L., Smith C.L., Westerfield M., Schofield P., Ruef B.J., Gkoutos G.V., Haendel M., Smedley D., Lewis S.E., and Robinson P.N.2014. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res.42(Database issue):D966–D974.
Kohler S., Schulz M.H., Krawitz P., Bauer S., Dolken S., Ott C.E., Mundlos C., Horn D., Mundlos S., and Robinson P.N.2009. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet.85(4):457–464.
Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J., Amin V., Whitaker J.W., Schultz M.D., Ward L.D., Sarkar A., Quon G., Sandstrom R.S., Eaton M.L., Wu Y.-C., Pfenning A., Wang X., ClaussnitzerYaping Liu M., Coarfa C., Alan Harris R., Shoresh N., Epstein C.B., Gjoneska E., Leung D., Xie W., David Hawkins R., Lister R., Hong C., Gascard P., Mungall A.J., Moore R., Chuah E., Tam A., Canfield T.K., Scott Hansen R., Kaul R., Sabo P.J., Bansal M.S., Carles A., Dixon J.R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G., Mercer T.R., Neph S.J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R.C., Siebenthall K.T., Sinnott-Armstrong N.A., Stevens M., Thurman R.E., Wu J., Zhang B., Zhou X., Abdennur N., Adli M., Akerman M., Barrera L., Antosiewicz-Bourget J., Ballinger T., Barnes M.J., Bates D., Bell R.J.A., Bennett D.A., Bianco K., Bock C., Boyle P., Brinchmann J., Caballero-Campo P., Camahort R., Carrasco-Alfonso M.J., Charnecki T., Chen H., Chen Z., Cheng J.B., Cho S., Chu A., Chung W.-Y., Cowan C., Athena Deng Q., Deshpande V., Diegel M., Ding B., Durham T., Echipare L., Edsall L., Flowers D., Genbacev-Krtolica O., Gifford C., Gillespie S., Giste E., Glass I.A., Gnirke A., Gormley M., Gu H., Gu J., Hafler D.A., Hangauer M.J., Hariharan M., Hatan M., Haugen E., He Y., Heimfeld S., Herlofsen S., Hou Z., Humbert R., Issner R., Jackson A.R., Jia H., Jiang P., Johnson A.K., Kadlecek T., Kamoh B., Kapidzic M., Kent J., Kim A., Kleinewietfeld M., Klugman S., Krishnan J., Kuan S., Kutyavin T., Lee A.-Y., Lee K., Li J., Li N., Li Y., Ligon K.L., Lin S., Lin Y., Liu J., Liu Y., Luckey C.J., Ma Y.P., Maire C., Marson A., Mattick J.S., Mayo M., McMaster M., Metsky H., Mikkelsen T., Miller D., Miri M., Mukame E., Nagarajan R.P., Neri F., Nery J., Nguyen T., O’Geen H., Paithankar S., Papayannopoulou T., Pelizzola M., Plettner P., Propson N.E., Raghuraman S., Raney B.J., Raubitschek A., Reynolds A.P., Richards H., Riehle K., Rinaudo P., Robinson J.F., Rockweiler N.B., Rosen E., Rynes E., Schein J., Sears R., Sejnowski T., Shafer A., Shen L., Shoemaker R., Sigaroudinia M., Slukvin I., Stehling-Sun S., Stewart R., Subramanian S.L., Suknuntha K., Swanson S., Tian S., Tilden H., Tsai L., Urich M., Vaughn I., Vierstra J., Vong S., Wagner U., Wang H., Wang T., Wang Y., Weiss A., Whitton H., Wildberg A., Witt H., Won K.-J., Xie M., Xing X., Xu I., Xuan Z., Ye Z., Yen C.-a., Yu P., Zhang X., Zhang X., Zhao J., Zhou Y., Zhu J., Zhu Y., Ziegler S., Beaudet A.E., Boyer L.A., De Jager P.L., Farnham P.J., Fisher S.J., Haussler D., Jones S.J.M., Li W., Marra M.A., McManus M.T., Sunyaev S., Thomson J.A., Tlsty T.D., Tsai L.-H., Wang W., Waterland R.A., Zhang M.Q., Chadwick L.H., Bernstein B.E., Costello J.F., Ecker J.R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J.A., Wang T., Kellis M., Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J., Amin V., Whitaker J.W., Schultz M.D., Ward L.D., Sarkar A., Quon G., Sandstrom R.S., Eaton M.L., Wu Y.-C., Pfenning A.R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R.A., Shoresh N., Epstein C.B., Gjoneska E., Leung D., Xie W., Hawkins R.D., Lister R., Hong C., Gascard P., Mungall A.J., Moore R., Chuah E., Tam A., Canfield T.K., Hansen R.S., Kaul R., Sabo P.J., Bansal M.S., Carles A., Dixon J.R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G., Mercer T.R., Neph S.J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R.C., Siebenthall K.T., Sinnott-Armstrong N.A., Stevens M., Thurman R.E., Wu J., Zhang B., Zhou X., Beaudet A.E., Boyer L.A., De Jager P.L., Farnham P.J., Fisher S.J., Haussler D., Jones S.J.M., Li W., Marra M.A., McManus M.T., Sunyaev S., Thomson J.A., Tlsty T.D., Tsai L.-H., Wang W., Waterland R.A., Zhang M.Q., Chadwick L.H., Bernstein B.E., Costello J.F., Ecker J.R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J.A., Wang T., and Kellis M.2015. Integrative analysis of 111 reference human epigenomes. Nature.518(7539):317–330.
Kutmon M., Riutta A., Nunes N., Hanspers K., Willighagen E.L., Bohler A., Melius J., Waagmeester A., Sinha S.R., Miller R., Coort S.L., Cirillo E., Smeets B., Evelo C.T., and Pico A.R.2016. WikiPathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res.44(D1):D488–D494.
Landrum M.J., Lee J.M., Benson M., Brown G., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Hoover J., Jang W., Katz K., Ovetsky M., Riley G., Sethi A., Tully R., Villamarin-Salomon R., Rubinstein W., and Maglott D.R.2016. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res.44(D1):D862–D868.
Lee D., Gorkin D.U., Baker M., Strober B.J., Asoni A.L., McCallion A.S., and Beer M.A.2015. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet.47(8):955–961.
Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., Tukiainen T., Birnbaum D.P., Kosmicki J.A., Duncan L.E., Estrada K., Zhao F., Zou J., Pierce-Hoffman E., Berghout J., Cooper D.N., Deflaux N., DePristo M., Do R., Flannick J., Fromer M., Gauthier L., Goldstein J., Gupta N., Howrigan D., Kiezun A., Kurki M.I., Moonshine A.L., Natarajan P., Orozco L., Peloso G.M., Poplin R., Rivas M.A., Ruano-Rubio V., Rose S.A., Ruderfer D.M., Shakir K., Stenson P.D., Stevens C., Thomas B.P., Tiao G., Tusie-Luna M.T., Weisburd B., Won H.H., Yu D., Altshuler D.M., Ardissino D., Boehnke M., Danesh J., Donnelly S., Elosua R., Florez J.C., Gabriel S.B., Getz G., Glatt S.J., Hultman C.M., Kathiresan S., Laakso M., McCarroll S., McCarthy M.I., McGovern D., McPherson R., Neale B.M., Palotie A., Purcell S.M., Saleheen D., Scharf J.M., Sklar P., Sullivan P.F., Tuomilehto J., Tsuang M.T., Watkins H.C., Wilson J.G., Daly M.J., and MacArthur D.G., and Exome Aggregation Consortium. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature.536(7616):285–291.
Li H. and Durbin R.2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics.26(5):589–595.
MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R., Adams D.R., Altman R.B., Antonarakis S.E., Ashley E.A., Barrett J.C., Biesecker L.G., Conrad D.F., Cooper G.M., Cox N.J., Daly M.J., Gerstein M.B., Goldstein D.B., Hirschhorn J.N., Leal S.M., Pennacchio L.A., Stamatoyannopoulos J.A., Sunyaev S.R., Valle D., Voight B.F., Winckler W., and Gunter C.2014. Guidelines for investigating causality of sequence variants in human disease. Nature.508(7497):469–476.
Maurano M.T., Haugen E., Sandstrom R., Vierstra J., Shafer A., Kaul R., and Stamatoyannopoulos J.A.2015. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet.47(12):1393–1401.
McCarthy D.J., Humburg P., Kanapin A., Rivas M.A., Gaulton K., Cazier J.B., and Donnelly P.2014. Choice of transcripts and software has a large effect on variant annotation. Genome Med.6(3):26.
McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., and DePristo M.A.2010. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res.20(9):1297–1303.
McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., and Cunningham F.2016. The Ensembl Variant Effect Predictor. Genome Biol.17(1):122.
Merico D., Roifman M., Braunschweig U., Yuen R.K., Alexandrova R., Bates A., Reid B., Nalpathamkalam T., Wang Z., Thiruvahindrapuram B., Gray P., Kakakios A., Peake J., Hogarth S., Manson D., Buncic R., Pereira S.L., Herbrick J.A., Blencowe B.J., Roifman C.M., and Scherer S.W.2015a. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat. Commun.6:8718.
Merico D., Sharfe N., Hu P., Herbrick J.-A., and Roifman C.M.2015b. RelB deficiency causes combined immunodeficiency. LymphoSign J.2(3):147–155.
Merico D., Zarrei M., Costain G., Ogura L., Alipanahi B., Gazzellone M.J., Butcher N.J., Thiruvahindrapuram B., Nalpathamkalam T., Chow E.W., Andrade D.M., Frey B.J., Marshall C.R., Scherer S.W., and Bassett A.S.2015c. Whole-genome sequencing suggests schizophrenia risk mechanisms in humans with 22q11.2 deletion syndrome. G3 (Bethesda).5(11):2453–2461.
Miller N.A., Farrow E.G., Gibson M., Willig L.K., Twist G., Yoo B., Marrs T., Corder S., Krivohlavek L., Walter A., Petrikin J.E., Saunders C.J., Thiffault I., Soden S.E., Smith L.D., Dinwiddie D.L., Herd S., Cakici J.A., Catreux S., Ruehle M., and Kingsmore S.F.2015. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med.7:100.
Mort M., Sterne-Weiler T., Li B., Ball E.V., Cooper D.N., Radivojac P., Sanford J.R., and Mooney S.D.2014. MutPred Splice: Machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol.15(1):R19.
Mu J.C., Tootoonchi Afshar P., Mohiyuddin M., Chen X., Li J., Bani Asadi N., Gerstein M.B., Wong W.H., and Lam H.Y.2015. Leveraging long read sequencing from a single individual to provide a comprehensive resource for benchmarking variant calling methods. Sci. Rep.5:14493.
Narayan S., Bader G.D., and Reimand J.2016. Frequent mutations in acetylation and ubiquitination sites suggest novel driver mechanisms of cancer. Genome Med.8(1):55.
Naseer M.I., Sogaty S., Rasool M., Chaudhary A.G., Abutalib Y.A., Walker S., Marshall C.R., Merico D., Carter M.T., Scherer S.W., Al-Qahtani M.H., and Zarrei M.2016. Microcephaly-capillary malformation syndrome: Brothers with a homozygous STAMBP mutation, uncovered by exome sequencing. Am. J. Med. Genet. A.170(11):3018–3022.
Ng P.C. and Henikoff S.2001. Predicting deleterious amino acid substitutions. Genome Res.11(5):863–874.
Ngan B., Merico D., Marcus N., Kim V.H.D., Upton J., Bates A., Herbrick J., Nalpathamkalam T., Thiruvahindrapuram B., Cox P., and Roifman C.M.2014. Mutations in tetratricopeptide repeat domain 7A (TTC7A) are associated with combined immunodeficiency with dendriform lung ossification but no intestinal atresia. LymphoSign J.1(1):10–26.
Noll A.C., Miller N.A., Smith L.D., Yoo B., Fiedler S., Cooley L.D., Willig L.K., Petrikin J.E., Cakici J., Lesko J., Newton A., Detherage K., Thiffault I., Saunders C.J., Farrow E.G., and Kingsmore S.F.2016. Clinical detection of deletion structural variants in whole-genome sequences. NPJ Genom. Med.1:16026.
Pabinger S., Dander A., Fischer M., Snajder R., Sperk M., Efremova M., Krabichler B., Speicher M.R., Zschocke J., and Trajanoski Z.2014. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform.15(2):256–278.
Palazzo A.F. and Lee E.S.2015. Non-coding RNA: What is functional and what is junk?Front. Genet.6:2.
Pang A.W., Macdonald J.R., Yuen R.K., Hayes V.M., and Scherer S.W.2014. Performance of high-throughput sequencing for the discovery of genetic variation across the complete size spectrum. G3 (Bethesda).4(1):63–65.
Petrovski S., Wang Q., Heinzen E.L., Allen A.S., and Goldstein D.B.2013. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet.9(8):e1003709.
Pollard K.S., Hubisz M.J., Rosenbloom K.R., and Siepel A.2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res.20(1):110–121.
Quek X.C., Thomson D.W., Maag J.L., Bartonicek N., Signal B., Clark M.B., Gloss B.S., and Dinger M.E.2015. lncRNAdb v2.0: Expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res.43(Database issue):D168–D173.
Ramu A., Noordam M.J., Schwartz R.S., Wuster A., Hurles M.E., Cartwright R.A., and Conrad D.F.2013. DeNovoGear: De novo indel and point mutation discovery and phasing. Nat. Methods.10(10):985–987.
Reese M.G., Moore B., Batchelor C., Salas F., Cunningham F., Marth G.T., Stein L., Flicek P., Yandell M., and Eilbeck K.2010. A standard variation file format for human genome sequences. Genome Biol.11(8):R88.
Reimand J., Wagih O., and Bader G.D.2013. The mutational landscape of phosphorylation signaling in cancer. Sci. Rep.3:2651.
Reva B., Antipin Y., and Sander C.2011. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res.39(17):e118.
Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., Voelkerding K., and Rehm H.L., and ACMG Laboratory Quality Assurance Committee. 2015. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med.17(5):405–424.
Rosenberg A.B., Patwardhan R.P., Shendure J., and Seelig G.2015. Learning the sequence determinants of alternative splicing from millions of random sequences. Cell.163(3):698–711.
Samocha K.E., Robinson E.B., Sanders S.J., Stevens C., Sabo A., McGrath L.M., Kosmicki J.A., Rehnström K., Mallick S., Kirby A., Wall D.P., MacArthur D.G., Gabriel S.B., DePristo M., Purcell S.M., Palotie A., Boerwinkle E., Buxbaum J.D., Cook E.H., Gibbs R.A., Schellenberg G.D., Sutcliffe J.S., Devlin B., Roeder K., Neale B.M., and Daly M.J.2014. A framework for the interpretation of de novo mutation in human disease. Nat. Genet.46(9):944–950.
Saunders C.J., Miller N.A., Soden S.E., Dinwiddie D.L., Noll A., Alnadi N.A., Andraws N., Patterson M.L., Krivohlavek L.A., Fellis J., Humphray S., Saffrey P., Kingsbury Z., Weir J.C., Betley J., Grocock R.J., Margulies E.H., Farrow E.G., Artman M., Safina N.P., Petrikin J.E., Hall K.P., and Kingsmore S.F.2012. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med.4(154):154ra135.
Scheper G.C., van der Knaap M.S., and Proud C.G.2007. Translation matters: Protein synthesis defects in inherited disease. Nat. Rev. Genet.8(9):711–723.
Scotti M.M. and Swanson M.S.2016. RNA mis-splicing in disease. Nat. Rev. Genet.17(1):19–32.
Sibley C.R., Blazquez L., and Ule J.2016. Lessons from non-canonical splicing. Nat. Rev. Genet.17(7):407–421.
Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Weinstock G.M., Wilson R.K., Gibbs R.A., Kent W.J., Miller W., and Haussler D.2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res.15(8):1034–1050.
Singleton M.V., Guthery S.L., Voelkerding K.V., Chen K., Kennedy B., Margraf R.L., Durtschi J., Eilbeck K., Reese M.G., Jorde L.B., Huff C.D., and Yandell M.2014. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am. J. Hum. Genet.94(4):599–610.
Smedley D. and Robinson P.N.2015. Phenotype-driven strategies for exome prioritization of human Mendelian disease genes. Genome Med.7(1):81.
Smedley D., Jacobsen J.O., Jager M., Kohler S., Holtgrewe M., Schubach M., Siragusa E., Zemojtel T., Buske O.J., Washington N.L., Bone W.P., Haendel M.A., and Robinson P.N.2015. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc.10(12):2004–2015.
Smedley D., Kohler S., Czeschik J.C., Amberger J., Bocchini C., Hamosh A., Veldboer J., Zemojtel T., and Robinson P.N.2014. Walking the interactome for candidate prioritization in exome sequencing studies of Mendelian diseases. Bioinformatics.30(22):3215–3222.
Smedley D., Schubach M., Jacobsen J.O., Kohler S., Zemojtel T., Spielmann M., Jager M., Hochheiser H., Washington N.L., McMurry J.A., Haendel M.A., Mungall C.J., Lewis S.E., Groza T., Valentini G., and Robinson P.N.2016. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in Mendelian disease. Am. J. Hum. Genet.99(3):595–606.
Smith C.L. and Eppig J.T.2012. The Mammalian Phenotype Ontology as a unifying standard for experimental and high-throughput phenotyping data. Mamm. Genome.23(9–10):653–668.
Solomon B.D., Nguyen A.D., Bear K.A., and Wolfsberg T.G.2013. Clinical genomic database. Proc. Natl. Acad. Sci. USA.110(24):9851–9855.
Soukarieh O., Gaildrat P., Hamieh M., Drouet A., Baert-Desurmont S., Frebourg T., Tosi M., and Martins A.2016. Exonic splicing mutations are more prevalent than currently estimated and can be predicted by using in silico tools. PLoS Genet.12(1):e1005756.
Stavropoulos D.J., Merico D., Jobling R., Bowdin S., Monfared N., Thiruvahindrapuram B., Nalpathamkalam T., Pellecchia G., Yuen R.K.C., Szego M.J., Hayeems R.Z., Shaul R.Z., Brudno M., Girdea M., Frey B., Alipanahi B., Ahmed S., Babul-Hirji R., Porras R.B., Carter M.T., Chad L., Chaudhry A., Chitayat D., Doust S.J., Cytrynbaum C., Dupuis L., Ejaz R., Fishman L., Guerin A., Hashemi B., Helal M., Hewson S., Inbar-Feigenberg M., Kannu P., Karp N., Kim R.H., Kronick J., Liston E., MacDonald H., Mercimek-Mahmutoglu S., Mendoza-Londono R., Nasr E., Nimmo G., Parkinson N., Quercia N., Raiman J., Roifman M., Schulze A., Shugar A., Shuman C., Sinajon P., Siriwardena K., Weksberg R., Yoon G., Carew C., Erickson R., Leach R.A., Klein R., Ray P.N., Meyn M.S., Scherer S.W., Cohn R.D., and Marshall C.R.2016. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. NPJ Genom. Med.1:15012.
Steinberg J., Honti F., Meader S., and Webber C.2015. Haploinsufficiency predictions without study bias. Nucleic Acids Res.43(15):e101.
Stenson P.D., Mort M., Ball E.V., Shaw K., Phillips A., and Cooper D.N.2014. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet.133(1):1–9.
Telenti A., Pierce L.T., Biggs W.H., di Iulio J., Wong E.H.M., Fabani M.M., Kirkness E.F., Moustafa A., Shah N., Xie C., Brewerton S.C., Bulsara N., Garner C., Metzker G., Sandoval E., Perkins B.A., Och F.J., Turpaz Y., and Venter J.C.2016. Deep sequencing of 10,000 human genomes.
Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Kang H.M., Jordan D., Leal S.M., Gabriel S., Rieder M.J., Abecasis G., Altshuler D., Nickerson D.A., Boerwinkle E., Sunyaev S., Bustamante C.D., Bamshad M.J., Akey J.M., Broad G.O., and Seattle G.O., and NHLBI Exome Sequencing Project. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science.337(6090):64–69.
Tranchevent L.C., Capdevila F.B., Nitsch D., De Moor B., De Causmaecker P., and Moreau Y.2011. A guide to web tools to prioritize candidate genes. Brief Bioinform.12(1):22–32.
Uddin M., Tammimies K., Pellecchia G., Alipanahi B., Hu P., Wang Z., Pinto D., Lau L., Nalpathamkalam T., Marshall C.R., Blencowe B.J., Frey B.J., Merico D., Yuen R.K., and Scherer S.W.2014. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat. Genet.46(7):742–747.
Vail P.J., Morris B., van Kan A., Burdett B.C., Moyes K., Theisen A., Kerr I.D., Wenstrup R.J., and Eggington J.M.2015. Comparison of locus-specific databases for BRCA1 and BRCA2 variants reveals disparity in variant classification within and among databases. J. Comm. Genet.6(4):351–359.
Veltman J.A. and Brunner H.G.2012. De novo mutations in human genetic disease. Nat. Rev. Genet.13(8):565–575.
Wagih O., Reimand J., and Bader G.D.2015. MIMP: Predicting the impact of mutations on kinase-substrate phosphorylation. Nat. Methods.12(6):531–533.
Wang K., Li M., and Hakonarson H.2010. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res.38(16):e164.
Wang X., Gulbahce N., and Yu H.2011. Network-based methods for human disease gene prediction. Brief Funct. Genomics.10(5):280–293.
Ware J.S., Samocha K.E., Homsy J., and Daly M.J.2015. Interpreting de novo variation in human disease using denovolyzeR. Curr. Protoc. Hum. Genet.87:7.25.1–7.25.15.
Wasserman W.W. and Sandelin A.2004. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet.5(4):276–287.
Weedon M.N.Cebola I.Patch A.M.Flanagan S.E.De Franco E.Caswell R.Rodriguez-Segui S.A.Shaw-Smith C.Cho C.H.Lango Allen H.Houghton J.A.Roth C.L.Chen R.Hussain K.Marsh P.Vallier L.Murray A., International Pancreatic Agenesis ConsortiumEllard S.Ferrer J.Hattersley A.T.2014. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat. Genet.46(1):61–64.
Wenger A.M., Clarke S.L., Guturu H., Chen J., Schaar B.T., McLean C.Y., and Bejerano G.2013. PRISM offers a comprehensive genomic approach to transcription factor function prediction. Genome Res.23(5):889–904.
Whalen S., Truty R.M., and Pollard K.S.2016. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet.48(5):488–496.
Xiong H.Y., Alipanahi B., Lee L.J., Bretschneider H., Merico D., Yuen R.K., Hua Y., Gueroussov S., Najafabadi H.S., Hughes T.R., Morris Q., Barash Y., Krainer A.R., Jojic N., Scherer S.W., Blencowe B.J., and Frey B.J.2015. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science.347(6218):1254806.
Xue Y., Chen Y., Ayub Q., Huang N., Ball E.V., Mort M., Phillips A.D., Shaw K., Stenson P.D., Cooper D.N., and Tyler-Smith C., and 1000 Genomes Project Consortium. 2012. Deleterious- and disease-allele prevalence in healthy individuals: Insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet.91(6):1022–1032.
Yang Y., Muzny D.M., Xia F., Niu Z., Person R., Ding Y., Ward P., Braxton A., Wang M., Buhay C., Veeraraghavan N., Hawes A., Chiang T., Leduc M., Beuten J., Zhang J., He W., Scull J., Willis A., Landsverk M., Craigen W.J., Bekheirnia M.R., Stray-Pedersen A., Liu P., Wen S., Alcaraz W., Cui H., Walkiewicz M., Reid J., Bainbridge M., Patel A., Boerwinkle E., Beaudet A.L., Lupski J.R., Plon S.E., Gibbs R.A., and Eng C.M.2014. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA.312(18):1870–1879.
Yeo G. and Burge C.B.2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol.11(2–3):377–394.
Yuen R.K., Merico D., Cao H., Pellecchia G., Alipanahi B., Thiruvahindrapuram B., Tong X., Sun Y., Cao D., Zhang T., Wu X., Jin X., Zhou Z., Liu X., Nalpathamkalam T., Walker S., Howe J.L., Wang Z., MacDonald J.R., Chan A., D’Abate L., Deneault E., Siu M.T., Tammimies K., Uddin M., Zarrei M., Wang M., Li Y., Wang J., Wang J., Yang H., Bookman M., Bingham J., Gross S.S., Loy D., Pletcher M., Marshall C.R., Anagnostou E., Zwaigenbaum L., Weksberg R., Fernandez B.A., Roberts W., Szatmari P., Glazer D., Frey B.J., Ring R.H., Xu X., and Scherer S.W.2016. Genome-wide characteristics of de novo mutations in autism. NPJ Genom. Med.1:160271–1602710.
Yuen R.K., Thiruvahindrapuram B., Merico D., Walker S., Tammimies K., Hoang N., Chrysler C., Nalpathamkalam T., Pellecchia G., Liu Y., Gazzellone M.J., D’Abate L., Deneault E., Howe J.L., Liu R.S., Thompson A., Zarrei M., Uddin M., Marshall C.R., Ring R.H., Zwaigenbaum L., Ray P.N., Weksberg R., Carter M.T., Fernandez B.A., Roberts W., Szatmari P., and Scherer S.W.2015. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med.21(2):185–191.
Zarrei M., MacDonald J.R., Merico D., and Scherer S.W.2015. A copy number variation map of the human genome. Nat. Rev. Genet.16(3):172–183.
Zemojtel T., Kohler S., Mackenroth L., Jager M., Hecht J., Krawitz P., Graul-Neumann L., Doelken S., Ehmke N., Spielmann M., Oien N.C., Schweiger M.R., Kruger U., Frommer G., Fischer B., Kornak U., Flottmann R., Ardeshirdavani A., Moreau Y., Lewis S.E., Haendel M., Smedley D., Horn D., Mundlos S., and Robinson P.N.2014. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci. Transl. Med.6(252):252ra123.
Zhang F. and Lupski J.R.2015. Non-coding genetic variants in human disease. Hum. Mol. Genet.24(R1):R102–R110.
Zhou J. and Troyanskaya O.G.2015. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods.12(10):931–934.
Zhu Y., Chen Z., Zhang K., Wang M., Medovoy D., Whitaker J.W., Ding B., Li N., Zheng L., and Wang W.2016. Constructing 3D interaction maps from 1D epigenomes. Nat. Commun.7:10812.
Zook J.M., Chapman B., Wang J., Mittelman D., Hofmann O., Hide W., and Salit M.2014. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol.32(3):246–251.
Zuberi K., Franz M., Rodriguez H., Montojo J., Lopes C.T., Bader G.D., and Morris Q.2013. GeneMANIA prediction server 2013 update. Nucleic Acids Res.41(Web Server issue):W115–W122.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 4 • December 2016
Pages: 135 - 158
History
Received: 26 September 2016
Accepted: 22 November 2016
Accepted manuscript online: 22 November 2016
Copyright
© 2016.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
DanieleMerico. 2016. Whole exome and genome sequencing for Mendelian immune disorders: from molecular diagnostics to new disease variant and gene discovery. LymphoSign Journal.
3(4): 135-158. https://doi.org/10.14785/lymphosign-2016-0011
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. A novel STAT3 splice-site variant in a kindred with autosomal dominant hyper IgE syndrome
2. A novel mutation in LIG4 in an infant presenting with severe combined immunodeficiency with thymic medullary dysplasia
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
