A clinical trial protocol for the study of intravenous immunoglobulin in patients with primary antibody immunodeficiency disorders
Abstract
This protocol has been excerpted from protocols recently used to study the safety, efficacy, and pharmacokinetics of 2 new intravenous immunoglobulins (IVIG). It incorporates the safety and efficacy parameters provided by the United States Food and Drug Administration in in a Guidance for Industry that contains recommendations for Investigational New Drug Application sponsors and Biologic License applicants for testing IVIG products as replacement therapy in primary humoral immunodeficiency. The format and contents of the protocol are generally the same as described in the ICH Guideline for Good Clinical Practice. We describe here the topics found in the main body of the protocol. We have omitted items that will be included in a formal protocol such as the title page, identifying number, name and contact information of the sponsor, names and contact information of the persons authorized to sign the protocol and amendments, etc. The success of a study of IVIG in patients with primary humoral immunodeficiency is very much dependent on the care in which patients are selected. It is extremely important to enroll patients who have been verified to be antibody deficient. Therefore, patient selection criteria should involve a thorough review of each patient’s infection history, serum IgG level prior to immunoglobulin therapy, and antibody responses to polysaccharide and protein vaccines.
Statement of novelty: This protocol is designed to select study subjects with well-documented antibody deficiencies and therefore will benefit from immunoglobulin therapy.
Introduction
Background: primary immunodeficiency diseases
Antibody deficiencies, also known as B-cell or humoral immunodeficiencies, comprise the largest proportion (up to 77%) of all primary immunodeficiency diseases (PID) (Yong et al. 2008). Antibody deficiency diseases result from an impaired ability to produce specific antibodies in response to antigens. Many antibody deficiencies are caused by mutations in genes involved in regulating B-cell differentiation or by mutations in the immunoglobulin genes themselves (Schroeder 2001).
X-linked agammaglobulinemia (XLA) is caused by a B lymphocyte differentiation defect known as the Bruton tyrosine kinase (BTK) deficiency (Bonilla and Geha 2003). XLA is an X-linked recessive disorder that affects men. Female carriers are generally asymptomatic (Yong et al. 2008). It is the most common genetic lesion identified in subjects lacking B cells and serum immunoglobulin. XLA subjects suffer from recurrent infections such as otitis media, sinusitis, conjunctivitis, pneumonia, and pyodema mainly due to Hemophilus influenzae and Streptococcus pneumonia (Rosen et al. 1995).
Common variable immunodeficiency (CVID) is the most common antibody deficiency disorder with an estimated prevalence in Europe and North America ranging from 1 in10,000 to 1 in 50,000 (Bergbreiter and Salzer 2009). CVID is characterized by low levels of serum immunoglobulin together with significant impairment of antibody responses to antigens introduced by vaccination or natural infection. CVID is a late-onset immunodeficiency with onset of symptoms in the third decade of life and diagnosis in the fourth decade of life (Bergbreiter and Salzer 2009). About 20% of subjects with CVID are diagnosed in childhood. CVID affects males and females equally (Bergbreiter and Salzer 2009).
Benefit/risk statement
Intravenous immunoglobulin (IVIG) therapy has been shown to prevent bacterial infections in subjects with primary antibody deficiencies. Reductions in hospitalizations and infections have been well documented in subjects receiving doses of >400 mg/kg every 21 or 28 days. Patients with chronic sinusitis and (or) pulmonary disease may benefit from higher doses (Roifman and Gelfand 1988). In general, maintenance of trough serum IgG levels above 500 mg/dL is a reasonable standard of care (Roifman and Gelfand 1988).
As with any medication, there are risks associated with the administration of IVIG. Acute adverse reactions may include headache, chills, nausea, flushing, and serious anaphylactic-like reactions. On rare occasions, aseptic meningitis, hemolysis, hyperviscosity, and thromboembolic events may occur (Pierce and Jain 2003). Since IVIG is a blood product, there is also a risk of transmission of blood-borne infections. There are no reports of HIV or hepatitis B transmission by IVIG but hepatitis C transmission was reported before current virus inactivation and removal methods were introduced (Pierce and Jain 2003).
Rationale
With the development of IVIG, doses have increased from the 100 to 200 mg/kg/month usually administered by intramuscular immune globulin (IMIG). The optimum dose and frequency of IVIG administration is determined individually for each subject. For subjects with PID, monthly doses of approximately 300–600 mg/kg infused every 21–28 days are common (Roifman and Gelfand 1988). Studies have shown that some patients, particularly those with chronic/recurrent sino-pulmonary disease, benefit from doses in the range of 800 mg/kg/dose (Roifman and Gelfand 1988; Eijkhout et al. 2001).
GCP compliance
The sponsor is committed to satisfying the requirements of the ICH Guideline for Good Clinical Practice, Title 21 of the Code of Federal Regulations, and the Declaration of Helsinki.
Study population
Subjects with well-defined PID that includes a major component of antibody deficiency will be studied.
Study objective
The objective of this study is to assess the safety, efficacy, and pharmacokinetics of an IVIG in subjects with PID.
Primary efficacy endpoint
The primary efficacy endpoint is the incidence of acute serious bacterial infections meeting United States Food and Drug Administration (FDA) criteria (bacterial pneumonia, bacteremia/sepsis, bacterial meningitis, visceral abscess, osteomyelitis/septic arthritis). The upper one-sided 99% confidence limit for the frequency of acute serious bacterial infections with the study IVIG must be less than 1.0 per subject per year (FDA/CBER 2008).
Secondary efficacy endpoints
•
The number of days missed work/school/kindergarten/day care or unable to perform normal daily activities due to infections, days of unscheduled physician visits and hospitalizations due to infection, days on therapeutic antibiotics, and incidence of infections other than acute serious bacterial infections.
Primary safety endpoint
•
The overall incidence of adverse events (AEs) that occur during or within 1 hour, 24 hours, and 72 hours following an infusion of test product, regardless of whether or not the AE is determined to be product related.
Secondary safety endpoints
•
All AEs that occur during the study regardless of the investigator’s assessment of their relationship to study product. Changes in vital signs, physical examination, and laboratory results. The proportion of IVIG infusions for which the infusion rate was decreased due to an AE. The proportion of AEs considered by the investigator to be product related. Viral safety (freedom from transmission of blood borne viral diseases).
Primary pharmacokinetic endpoints
•
Pharmacokinetic (PK) parameters of total IgG in up to 20 patients.
•
Trough serum total IgG levels before each infusion of IVIG in all subjects and the interval between infusions will be recorded.
Secondary pharmacokinetic endpoints
•
PK parameters of IgG subclasses and specific IgG antibodies:
◦
anti-Hemophilus influenza type b
◦
anti-Streptococcus pneumonia serotypes
◦
anti-Tetanus toxoid
◦
anti-measles
Investigational plan
This will be a prospective, open label, single-arm, multicenter, historically controlled, Phase III study.
The study design is an open-label, uncontrolled study of at least 40 evaluable subjects with well-defined PID and antibody deficiency. To guard against dropout, up to 50 subjects will be enrolled. All subjects will be followed for 12 months during which infusions will be given every 21 or 28 days. To demonstrate efficacy, the serious bacterial infection rate will be required to have an upper one-sided 99% confidence limit less than 1.0 per subject per year. Pharmacokinetic samples will be determined before and after the fifth study infusion.
Duration of the study
The study period for each subject is expected to be 14 months including up to 28 days for screening prior to the first study infusion, 12 months of study infusions, and a follow-up visit at 21 or 28 days after the last study infusion.
The total study duration will be approximately 25 months: trial set-up, 2 months; enrollment, 6 months; treatment and follow-up, 14 months; close-out: 3 months.
Investigators and study centers
A sufficient number of study sites to enroll up to 50 subjects will be recruited. Subjects who withdraw (or are withdrawn) will not be replaced.
Randomization and stratification
Not applicable.
Blinding
Not applicable.
Selection of study population
Subjects with a confirmed diagnosis of primary humoral immunodeficiency will be selected. Confirmation of PID diagnosis will include a review of infection history, serum IgG concentration at diagnosis and prior to immunoglobulin therapy and responses to vaccination with polysaccharide and protein vaccines.
Inclusion criteria
1.
Subjects with a confirmed clinical diagnosis of a PID as defined by IUIS (International Union of Immunological Societies 1999) and require treatment with IVIG. Documented agammaglobulinemia or hypogammaglobulinemia (preferably with documented antibody deficiency).
2.
Male or female, ages 2–70 years at time of screening.
3.
The subject has received 300–900 mg/kg of licensed IVIG therapy at 21 or 28 day intervals for at least 3 months prior to this study.
4.
At least 2 documented IgG trough levels of ≥5 g/L are obtained at 2 infusion cycles within 12 months prior to study enrollment.
5.
Subject is willing to comply with all requirements of the protocol.
6.
Females of child-bearing potential with a negative urine pregnancy test and who agree to employ adequate birth control measures during the study.
7.
Subject, parent, or guardian has signed the informed consent form and a child assent form if appropriate. Pediatric subjects are defined as 2–17 years of age at study entry and will require assent forms as appropriate per study documentation and regulations of the local jurisdiction.
8.
Authorization to access personal health information.
9.
Subjects currently participating in a clinical trial with an unlicensed IVIG may be enrolled if they received stable IVIG therapy for at least 3 infusion cycles prior to receiving the study IVIG and all inclusion and exclusion criteria are satisfied.
10.
Other IVIGs will be prohibited 1 infusion cycle (21 or 28 days) prior to the first infusion of study IVIG and until 1 infusion cycle after the last infusion of study IVIG.
11.
Subjects currently participating in a trial of subcutaneous immunoglobulin (SCIG) can be enrolled if they are switched to IVIG for three 21 or 28 day infusion cycles prior to enrollment in this study.
Exclusion criteria
1.
Subject has secondary immunodeficiency.
2.
Subject is newly diagnosed and has not been treated with immunoglobulin or has been diagnosed with dysgammaglobulinemia or isolated IgG subclass deficiency.
3.
Subject has a history of repeated reactions or hypersensitivity to IVIG and other injectable forms of IgG.
4.
Subject has a history of thrombotic events including deep vein thrombosis, cerebrovascular accident, pulmonary embolism, transient ischemic attacks, or myocardial infarction, as defined by at least 1 event in the subject’s lifetime.
5.
Subject has IgA deficiency and is known to have antibodies to IgA.
6.
Subject has received blood products other than human albumin or human immunoglobulin within 12 months prior to enrollment.
7.
Subject has significant protein losing enteropathy, nephrotic syndrome, or lymphangiectasia.
8.
Subject has an acute infection as documented by culture or diagnostic imaging and (or) a body temperature exceeding 38.5 °C (101.3 °F) within 7 days prior to screening.
9.
Subject has a known history, or is positive at enrollment, for human immunodeficiency virus (HIV) type 1/2 by NAT, hepatitis B virus (HBsAg or NAT), hepatitis C virus (by NAT), or hepatitis A virus (by NAT).
10.
Subject has levels of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) >2.5 times of the upper limit of normal from the laboratory designated for the study.
11.
Subject is using an implanted venous access device.
12.
Subject has profound anemia or persistent severe neutropenia (≤1000 neutrophils per mm3).
13.
Subject has a severe chronic condition such as renal failure (creatinine concentration >2.0 times the upper limit of normal) with proteinuria, congestive heart failure (New York Heart Association III/IV), cardiomyopathy, cardiac arrhythmia associated with thromboembolic events (e.g., atrial fibrillation), unstable or advanced ischemic heart disease, hyperviscosity, or any other condition that the investigator believes is likely to interfere with evaluation of the study drug or with satisfactory conduct of the trial.
14.
Subject has a history of a malignant disease other than properly treated carcinoma in situ of the cervix or basal cell or squamous cell carcinoma of the skin within 24 months prior to enrollment.
15.
Subject has history of epilepsy or migraines not completely controlled by medication.
16.
Subject is receiving the following medication:
a.
Steroids (oral or parenteral daily dose of ≥0.15 mg/kg/day of prednisone or equivalent).
b.
Other immunosuppressive drugs or chemotherapy.
17.
Females who are pregnant, breast feeding, or planning a pregnancy during the course of the study. Women who become pregnant during the study will be withdrawn from the study.
18.
Subject has participated in another clinical study within 3 weeks prior to study enrollment.
Withdrawal of subjects
A subject can withdraw from the trial at any time, or a parent may withdraw a child who is a subject, without prejudice. The reason for withdrawal for all subjects who do not complete the 12-month treatment period, including those who are withdrawn after enrollment but prior to their first infusion, will be recorded.
The investigator may withdraw a subject from the study in the following cases:
•
For safety reasons, such as a severe or serious AE, that do not justify continuation in the study in the opinion of the investigator;
•
For a protocol violation that jeopardizes performance of the study;
•
The subject does not comply with the protocol;
•
Continued participation will pose a risk to the subject;
•
Pregnancy;
•
Use of other IgG products;
•
Use of hyperimmune serum.
Early discontinuation of study
During the study, if safety concerns arise that indicate the study should be stopped, the sponsor will terminate the study (21CFR 312.56(d)).
Safety concerns that will be evaluated by the sponsor and the Data Safety Monitoring Board will include:
•
Serious, life-threatening, or unexpected adverse drug experiences that are clearly related to the investigational product.
•
Suspected thrombotic events.
IND Safety Reports will be submitted to the FDA according to 21CFR 312.32 requirements and the requirements of Health Canada.
In addition, conditions that may warrant study termination at a particular site include, but are not limited to, the following (21CFR 312.56(b)):
•
The discovery of unexpected and significant or unacceptable risks for the subjects in the study, usually arising due to GCP violations;
•
A decision of the sponsor to suspend the site for enrollment until all GCP issues have been addressed.
If the trial is prematurely terminated or suspended, the sponsor will inform the investigators, the Institutional Review Boards (IRB), and the FDA promptly of the termination or suspension and the reason(s) for the termination or suspension.
Study conduct
Treatment regimens
All subjects will receive intravenous infusions of study product at the same dose and interval as their previous IVIG therapy. Study IVIG will be administered at a dose of 0.3–0.9 g/kg (of body weight) every 21 or 28 days (±4 days) for a period of 12 months.
The dose regimen will remain unchanged throughout the study period unless there is a medically justified need to change it. Subjects can be treated with different lots of study IVIG over the course of the 12-month treatment period. Different lots may not be mixed for a single infusion.
Treatment assignment
After a subject has supplied written informed consent/assent and has fulfilled all inclusion and exclusion criteria of the trial, the subject will be enrolled in the study.
Blood samples required for PK analysis will be taken before and after the fifth study infusion for those subjects that consent to the PK portion of the study.
Subject identification
Subjects entering the study will be assigned subject identifiers: subject’s initials (first/middle/last name) and a unique subject number. The unique subject ID number will be in the format xx-yy composed of the study center number (xx) and subject number (yy). The center number will be assigned by the sponsor. The subject number will be assigned sequentially by the investigator beginning with 01.
Physical examination
A physical examination will be performed at screening and at each infusion visit. The general physical examination will include an evaluation of all body systems as the normal standard of care at each site. Information about the physical examination must be present in the source documentation at the study site.
Medical history and demography
A complete medical history, especially history of PID diagnostic test results, immunoglobulin treatment, and demography will be collected at the screening visit and recorded in case report forms (CRF). Subjects must have a documented history of stable IVIG therapy at 21 or 28 day intervals for at least 3 months prior to receiving IVIG. There must also be documentation of at least 2 trough serum levels of ≥5 g/L during the previous 12 months.
Subject diary
Subjects will be given a diary at the first infusion visit. The investigator will explain that the diary is a very important study document and that entries should be made daily to collect all safety and efficacy data. Information entered since the previous visit will be reviewed at every visit. The subject will enter the following information in the diary:
•
All AEs that occurred after the previous study infusion with special emphasis on AEs during the first 24, 48, and 72 hours after infusion.
•
Infections of any type.
•
Any medication taken, prescription and non-prescription.
•
The number of days of missed work/school/kindergarten/day care or inability to perform normal activities due to an infection.
•
The number of days of unscheduled physician visits and days of hospitalization due to an infection.
Subjects will be asked additional questions about their well-being in between their clinic visits to ensure all necessary information is recorded. Any discrepancies between the subject diaries and source notes will be provided as supplemental data on the CRF.
Dosage
The amount of IVIG in mg/kg to be administered is to be calculated by the investigator from the body weight to maintain a trough level of ≥5 g/L. The subject’s weight will be measured and recorded at screening and before every infusion to determine if a dose adjustment is required. Deviations in body weight of ±10% will not require a dose adjustment. Deviations in excess of ±10% since the subject’s previous infusion may require dose adjustment at the discretion of the investigator.
The number of vials for one infusion will be recorded in the investigator and (or) Pharmacy File.
Visit schedule
Table 1 summarizes the timeline for the study. Each study visit will be allowed a window of ±4 days.
Table 1:
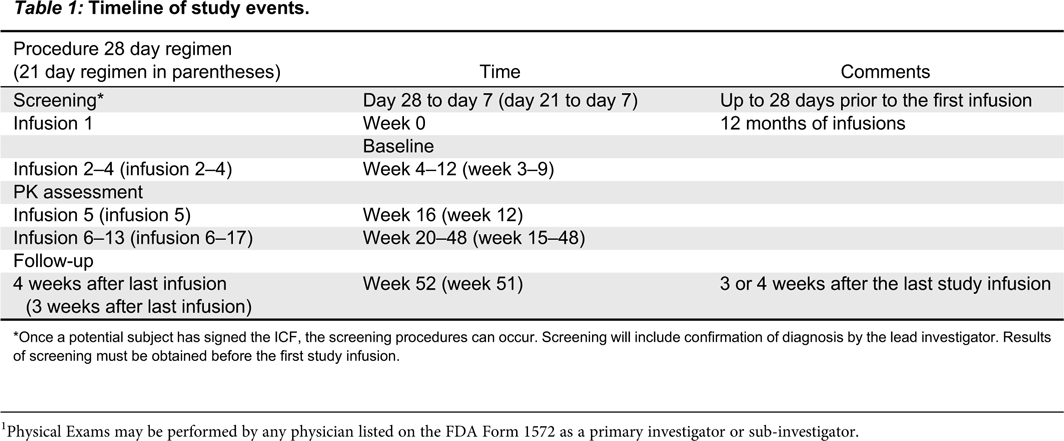
*
Once a potential subject has signed the ICF, the screening procedures can occur. Screening will include confirmation of diagnosis by the lead investigator. Results of screening must be obtained before the first study infusion.
Screening: day 28 or day 21 to day 7
•
Administer informed consent, and the authorization to access personal health information (and assent if applicable).
•
Review medical history and confirm diagnosis of primary immunodeficiency: infection history, date of PID diagnosis, IgG level before IVIG therapy, antibody responses to vaccination when available.
•
Meets inclusion/exclusion criteria.
•
Demography, physical examination, body weight, height, vital signs1.
•
Document IVIG therapy for at least 3 months prior to enrollment.
•
Ensure that 2 IgG trough levels of ≥5 g/L are documented within 12 months prior to enrollment.
•
Obtain chest X-ray (unless X-ray taken within previous 6 months is available to be used as baseline).
•
Check vital signs.
•
Obtain samples for viral marker testing: serology and NAT for HIV-1/2, HCV, HBV, HAV, B19, and a 1.0 mL retention sample to be stored at −70 °C.
•
•
Urinalysis (includes a pregnancy test for women of child bearing potential)4.
•
Confirm contraceptive use for women of child bearing potential.
First infusion (day 0, week 0)
Pre-infusion
•
Review screening test results and confirm that all are satisfactory.
•
Take vital signs.
•
Physical exam, body weight.
•
Ensure that subject is not volume depleted, i.e., is well-hydrated.
•
Take samples for blood chemistry, hematology sample, DAT/Coombs, IgG, IgG subclasses, specific antibodies, and retention.
•
Urinalysis sample.
•
Record concomitant medications.
•
Supply subject with diary and review instructions for completion.
During infusion
•
Record vital signs.
•
Check for AEs.
Post-infusion
•
Record vital signs.
•
DAT/Coombs blood sample.
•
Check for AEs within 1, 24, and 72 hours after the end of infusion.
◦
Study site will call the subject to ask open ended questions about their well-being and also to ensure that AEs are being documented.
Second infusion to final study infusion
Pre-infusion
•
Vital signs.
•
Physical exam, body weight.
•
Ensure that subject is not volume depleted, i.e., is well-hydrated.
•
Take samples for blood chemistry, hematology, IgG trough level, and retention.
•
Urinalysis sample.
•
Review subject diary and record entries, AEs.
•
Record concomitant medications.
During infusion
•
Vital signs.
•
Record AEs.
Post-infusion
•
Vital signs.
•
Record AEs 1, 24, and 72 hours after the end of infusion.
◦
Study site will call the subject to ask open ended questions about their well-being and also to ensure that AEs are being documented.
Additional tests
28-day infusion cycle
•
Urinalysis: infusions 3–13 and follow-up.
•
Viral safety: infusions 4, 7 and follow-up.
•
IgG subclasses, specific antibodies trough levels; before infusions 5 (non-PK subjects), 9, 13 (all subjects).
•
Direct Coombs infusions 6, 13 (before and after infusion).
21-day infusion cycle
•
Urinalysis: infusions 3–17 and follow-up.
•
Viral safety: infusions 5, 9, and follow-up.
•
IgG subclasses, specific antibodies trough levels: before infusions 5 (non-PK subjects) 11, and 17 (all subjects).
•
Direct Coombs: infusions 8 and 17 (before and after infusion).
Follow-up
•
Physical exam, weight.
•
Vital signs.
•
Samples for blood chemistry, hematology, IgG trough level, and retention.
•
Urinalysis.
•
Record AEs.
•
Review subject diary and record entries.
•
Record concomitant medications.
Unscheduled visit procedures
•
Interim visits related to the subject’s PID should be recorded in the CRF with any pertinent medical findings.
Study drug preparation and handling
The study IVIG should be a clear or slightly opalescent, colorless solution. Do not use if the solution is cloudy, turbid, or contains particulates.
The vial must not be shaken because shaking will cause foaming and loss of biological activity.
Do not freeze and do not use any IVIG solution that has been frozen.
Administer IVIG at room temperature.
Do not use study IVIG after expiration date.
The vial is for single use only. The product contains no preservative. Once the vial has been entered under aseptic conditions, its contents should be used promptly.
Infuse the study product using a separate infusion line. Do not mix with other intravenous medications or other IVIG products. An infusion pump may be used to control the rate of administration. For large doses, several vials may be pooled in a sterile infusion bag using aseptic technique. However, vials from different lots may not be combined.
The study drug will be administered intravenously. Prior to infusion, the investigator or his/her delegate must ensure that the subject is adequately hydrated.
At the end of an infusion, the tubing may be flushed with saline or D5W solution to ensure that the entire dose is administered. The infusion set may be primed with saline or D5W solution to fill the dead space. Once the infusion set is primed the infusion should begin immediately.
Infusion rates
The first dose of study IVIG should be infused at an initial rate as shown in Table 2.
Table 2:
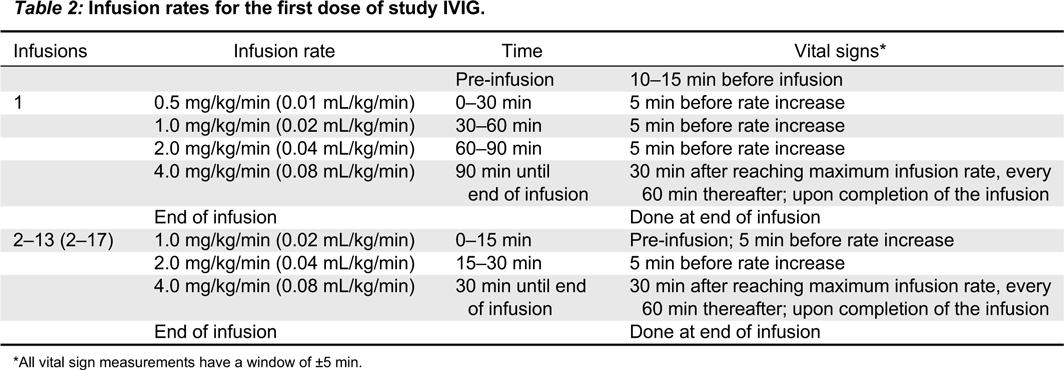
*
All vital sign measurements have a window of ±5 min.
Vital signs (heart rate, respiration rate, diastolic and systolic blood pressure, and body temperature) will be recorded as shown in Table 2.
If the first infusion is well-tolerated, the second study dose may be started at an initial infusion rate of 1.0 mg/kg/min (0.02 mL/kg/min) and increased every 15 min to the maximum rate of 4.0 mg/kg/min (0.08 mL/kg/min).
Subjects should be monitored for AEs during the infusion since there have been reports of thrombotic events and non-cardiogenic pulmonary edema (transfusion-related acute lung injury (TRALI)). If TRALI is suspected, appropriate tests will be performed for the presence of antineutrophil antibodies in both the product and the subject serum.
Product description
TBD.
Labeling and packaging
TBD.
Shipment and storage
The study IVIG will be shipped and stored at 2–8 °C.
Concomitant medications
Subjects who are prone to AEs associated with IVIG infusions are often premedicated with antihistamines, antipyretics, antibiotics and (or) steroids. Premedication intended to ameliorate AEs associated with infusion of study IVIG should be avoided in this study, if possible.
Subjects may receive premedication with acetyl salicylic acid (e.g., aspirin), nonsteroidal anti-inflammatory drugs (NSAIDS), or corticosteroids if they previously received such premedication while receiving treatment with another IVIG product. The subject’s previous use of premedication with other IVIG products and the AE for which the premedication was intended to prevent will be recorded. Other agents, e.g., antihistamines or acetaminophen, may be used when pre-infusion medication is considered to be necessary by the investigator.
The criteria used to initiate premedication to prevent re-occurrence of AEs will be recorded.
Any medication(s) (excluding biologics or chemotherapy agents) taken by the subject on a regular basis may be continued. In particular, daily low dose (≤100 mg) aspirin taken for cardiovascular prophylaxis, arthritis medications (including higher dose aspirin), and salicylates for inflammatory bowel disease may be continued.
Concomitant medications (including over-the-counter medications) with start and stop dates must be recorded on the subject’s CRF. The indication for treatment must be recorded including drug name, route and duration of drug administration. Concomitant medications will be recorded starting on the first day of infustion until the first follow-up visit.
Prohibited medications
Other IVIGs will be prohibited after the first infusion of study IVIG. High dose steroids (oral and parenteral, daily ≥0.15 mg of prednisone equivalent/kg/day) and other immunosuppressive or cytotoxic drugs are prohibited during the study except when required for emergency use. These medications must be recorded in the CRF.
Investigational product accountability and documentation
Investigational product will be stored and administered at the investigational site by trained personnel. Each vial will have a lot number and a unique vial number. The vials will arrive in boxes containing multiple vials. These boxes can be discarded or destroyed upon receipt and reconciliation of the IVIG vials contained within. An Investigational Product Accountability Record for the IVIG used for this study must be kept current by the clinical site and must contain:
•
Dates, quantities, expiration dates, and lot number(s) of all investigational product received.
•
Dates, quantities, vial numbers, and lot number(s) of investigational product dispensed for each infusion for each subject.
•
Subject number and initials.
•
Initials of the staff person dispensing the product.
The investigator must account for all product and supplies used in the study. At the end of the study, a final investigational product reconciliation statement must be completed by each site.
Inventory records must be readily available for inspection by the trial monitor and (or) auditor, and open to Regulatory Authority inspection or sponsor/CRO inspection.
The sponsor and the pharmacist (or otherwise dedicated person) must keep records regarding shipment, receipt, storage temperature logs, distribution, drug accountability, and destruction of the study medication.
Destruction of used and unused study drug
Any drug labels, empty vials, and partly used or unused vials must not be destroyed until the inventory records have been checked by the monitor. After verification of the inventory documentation, the monitor will inform the pharmacist (or otherwise dedicated person) to destroy empty and partially used vial at the site. At the end of the study, any remaining used and unused vials may be destroyed at site (upon confirmation by the monitor).
Assessment of efficacy
Primary Efficacy Endpoint: Incidence of the following acute serious bacterial infections: Bacteremia/sepsis, Bacterial meningitis Osteomyelitis/Septic Arthritis, Bacterial Pneumonia, Visceral Abscess
The diagnostic criteria for each acute serious bacterial infections are listed in the FDA Guidance for Industry (FDA/CBER, 2008).
Secondary efficacy endpoints
The subjects will record the following efficacy information in the diary that will be provided at the first infusion visit. Information entered since the previous visit will be reviewed at every visit.
•
Days missed from work, school, kindergarten, day care, or days unable to perform normal activities because of infection.
•
Infections other than serious acute bacterial infections.
•
Days of unscheduled physician visits and hospitalizations.
Other secondary efficacy endpoints include:
•
Number of days on therapeutic antibiotics.
The number and proportion of subjects who receive therapeutic antibiotics, the number of separate occasions, and the number of days they are taken during the study period will be recorded.
Assessment of pharmacokinetics
•
IgG trough levels.
Blood samples for the preparation of serum will be collected from all subjects at screening, prior to each infusion and at the follow-up visit to determine the trough levels of total IgG.
•
IgG subclasses/specific antibodies—trough levels.
28-day infusion subjects—IgG subclasses, specific antibodies trough levels: before infusions 5 (non-PK subjects), 9, and 13 (all subjects).
21-day infusion subjects—IgG subclasses, specific antibodies trough levels: before infusions 5 (non-PK subjects) 11, and 17 (all subjects).
•
Pharmacokinetic parameters of total IgG and serum levels of IgG subclasses and specific IgG antibodies.
Blood samples will be collected from up to 20 subjects before and after the fifth infusion of investigational product. Serum concentrations of total IgG, IgG subclasses, and specific antibodies will be determined. The pharmacokinetics of total IgG will be calculated.
The following pharmacokinetic parameters for total IgG will be determined using uncorrected values and baseline corrected values if IgG levels prior to immunoglobulin therapy are available.
•
Plasma concentration-time curve.
•
Half life.
•
Area under the curve (AUC0-t, AUC0-inf).
•
Maximum concentration (Cmax).
•
Minimum (trough) concentration (Cmin).
•
Time of maximum concentration (Tmax).
•
Clearance (CL), dose adjusted, e.g., mL/h/kg.
•
IR (incremental recovery).
•
Vz-terminal exponential volume of distribution (weight adjusted, mL/kg).
•
Vss-volume of distribution at steady state (weight adjusted).
•
Mean residence time (MRT).
•
% AUC ext-% of AUC from last data point to infinity as a percentage of AUC0-inf.
The following pharmacokinetic parameters for IgG subclasses and specific IgG antibodies will be calculated: Cmax, Cmin, and t1/2.
PK blood samples will be obtained 30 min to 10 min pre-infusion; 30 min (±5 min) and 2 hours (±15 min) post-infusion; and 24 hours (±2 hours), 3 days (±1 day), 7 days (±1 day), 14 days (±1 day), and 21 (±1 day) and 28 days (±2 days), (if applicable) post infusion.
The subjects must have given informed consent to participate in these assessments.
The samples for total IgG pharmacokinetics and the samples for IgG subclasses and specific antibodies will be analyzed by the central laboratory.
Assessment of safety
Safety parameters
Vital signs
Vital signs, including systolic and diastolic blood pressure, heart rate, respiratory rate, and body temperature will be recorded at screening, before, during, and after each infusion, and at the follow-up visit. Temperature should be measured either sublingually or auricularly and the method should be consistent throughout the study for a given subject. All vital sign measurements will have an allowable window of ±5 min. For each infusion, vital signs will be measured as previously described (Tables 3 and 4).
Table 3:
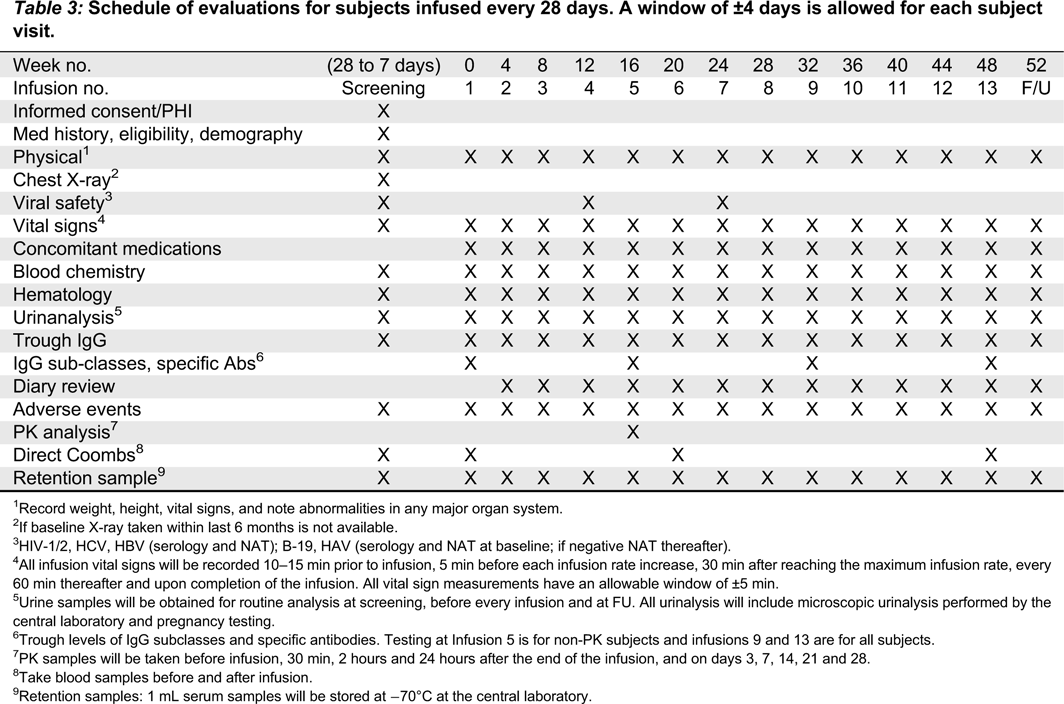
1
Record weight, height, vital signs, and note abnormalities in any major organ system.
2
If baseline X-ray taken within last 6 months is not available.
3
HIV-1/2, HCV, HBV (serology and NAT); B-19, HAV (serology and NAT at baseline; if negative NAT thereafter).
4
All infusion vital signs will be recorded 10–15 min prior to infusion, 5 min before each infusion rate increase, 30 min after reaching the maximum infusion rate, every 60 min thereafter and upon completion of the infusion. All vital sign measurements have an allowable window of ±5 min.
5
Urine samples will be obtained for routine analysis at screening, before every infusion and at FU. All urinalysis will include microscopic urinalysis performed by the central laboratory and pregnancy testing.
6
Trough levels of IgG subclasses and specific antibodies. Testing at Infusion 5 is for non-PK subjects and infusions 9 and 13 are for all subjects.
7
PK samples will be taken before infusion, 30 min, 2 hours and 24 hours after the end of the infusion, and on days 3, 7, 14, 21 and 28.
8
Take blood samples before and after infusion.
9
Retention samples: 1 mL serum samples will be stored at −70°C at the central laboratory.
Table 4:
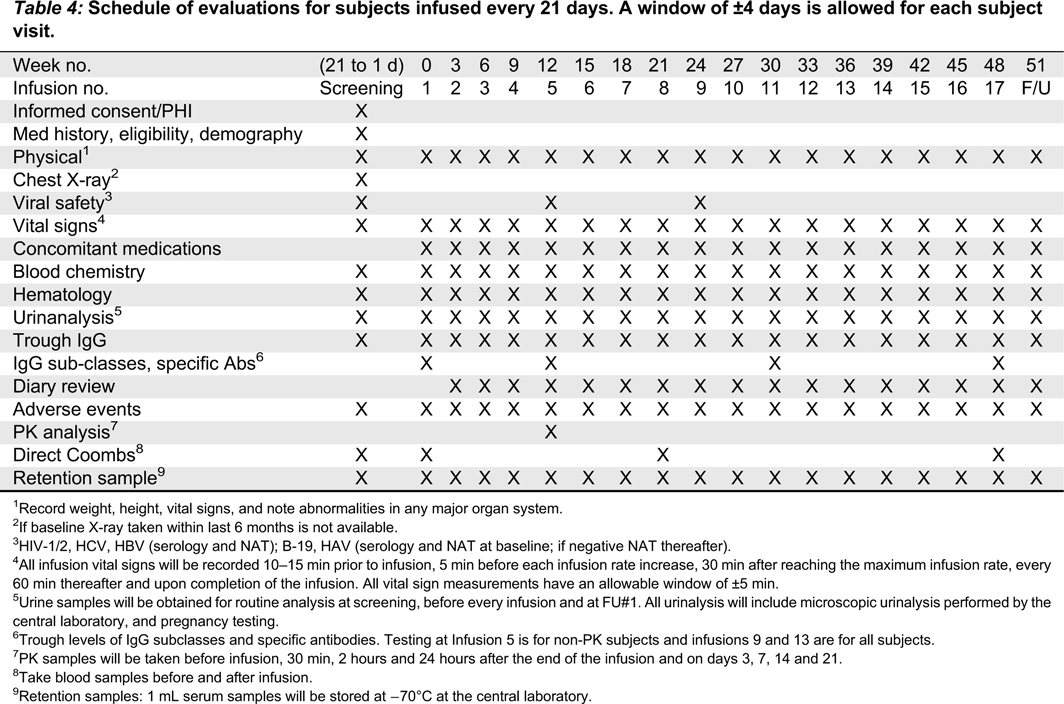
1
Record weight, height, vital signs, and note abnormalities in any major organ system.
2
If baseline X-ray taken within last 6 months is not available.
3
HIV-1/2, HCV, HBV (serology and NAT); B-19, HAV (serology and NAT at baseline; if negative NAT thereafter).
4
All infusion vital signs will be recorded 10–15 min prior to infusion, 5 min before each infusion rate increase, 30 min after reaching the maximum infusion rate, every 60 min thereafter and upon completion of the infusion. All vital sign measurements have an allowable window of ±5 min.
5
Urine samples will be obtained for routine analysis at screening, before every infusion and at FU#1. All urinalysis will include microscopic urinalysis performed by the central laboratory, and pregnancy testing.
6
Trough levels of IgG subclasses and specific antibodies. Testing at Infusion 5 is for non-PK subjects and infusions 9 and 13 are for all subjects.
7
PK samples will be taken before infusion, 30 min, 2 hours and 24 hours after the end of the infusion and on days 3, 7, 14 and 21.
8
Take blood samples before and after infusion.
9
Retention samples: 1 mL serum samples will be stored at −70°C at the central laboratory.
Routine blood tests
Blood samples for routine chemistry and hematology testing will be obtained at the screening visit, before each infusion, and at the follow-up visit.
The following parameters will be measured:
Hematology: hemoglobin, hematocrit, platelets, red blood cells (RBC), white blood cells (WBC), and differential counts.
Blood chemistry: total bilirubin, creatinine, blood urea nitrogen (BUN), ALT, AST, alkaline phosphatase, lactate dehydrogenase (LDH), glucose, sodium, potassium, chloride, CO2, and calcium.
Urinalysis tests
Urinalysis at all time points specified below will include microscopic examination of urine sediment and a pregnancy test.
If the subject is female, study personnel should ask for confirmation of contraceptive use at each infusion visit.
28-day infusion subjects: urinalysis will occur at screening, infusions 1–13, and follow-up.
21-day Infusion Subjects: urinalysis-will occur at screening, infusions 1–17, and follow-up.
Virology
Blood samples will be obtained at screening to exclude subjects who are positive by NAT for any of the following viruses: HIV, HCV, HBV, and HAV. A pretreatment serum sample will be retained and stored at −70 °C for possible future testing. Blood samples will also be collected at week 12 (infusion 4 in 28-day subjects; infusion 5 in 21-day subjects), week 24 (infusion 7 in 28-day subjects; infusion 9 in 21-day subjects), and the follow-up visit (21 or 28 days after the last study infusion).
The samples will be tested by serology and NAT for HIV-1/2, HCV, and HBV. Tests for HAV and B19 will be performed by NAT and serology (only at baseline). All viral safety measurements will be performed by the central laboratory.
Direct Coombs test
Measurements of DAT/Coombs will be performed before and after the first infusion, before and after infusion 5 and at the follow-up visit.
If a Direct Coombs test is positive after a study infusion, the test will be repeated and RBC count, hematocrit, hemoglobin, serum haptoglobin, bilirubin (total, direct, and indirect), LDH, plasma-free hemoglobin, and urine hemosiderin tests will be performed within 2–5 days after the infusion (all testing performed in response to a positive Direct Coombs will be performed by the central lab, including the repeat Direct Coombs test). Antibodies will be eluted from the RBCs and their specificity will be determined. The subject will be assessed for clinically significant hemolysis by testing for plasma hemoglobin. A drop in plasma hemoglobin of 2 g/L or more together with a drop in serum haptoglobin and a rise in LDH is indicative of intravascular hemolysis.
Retention samples
Retention serum samples will be obtained every visit from screening to the follow-up visit, and they will be stored at −70 °C at the central reference laboratory should additional tests be required.
Adverse events
Definition of an AE
An AE is defined as any treatment emergent unfavorable and unintended sign or symptom (including abnormal laboratory findings) that occurs at any time after the subject has signed informed consent until the first follow-up visit after the last study infusion, whether or not considered to be drug related. These will be recorded as AEs in the CRF.
Adverse reaction
An adverse reaction is any AE caused by the investigational product.
Suspected adverse reaction
A suspected adverse reaction is defined as any AE for which there is a reasonable possibility that the investigational product caused the AE. For the purposes of IND safety reporting, “reasonable possibility” means there is evidence to suggest a causal relationship between the drug and the AE.
Unexpected AEs
An AE or suspected adverse reaction is considered “unexpected” if it is not listed in the investigator brochure or is not listed at the specificity or severity that has been observed. Unexpected, as used in this definition, also refers to AEs or suspected adverse reactions that are mentioned in the investigator brochure as occurring with a class of drugs or as anticipated from the pharmacological properties of the drug, but are not specifically mentioned as occurring with the particular drug under investigation.
Serious adverse event (SAE)
A SAE is any adverse event that:
•
Results in death.
•
Is life-threatening.
•
Requires inpatient hospitalization or prolongation of existing hospitalization (does not include hospitalizations for elective procedures for pre-existing conditions that did not worsen from baseline).
•
Results in persistent or significant disability/incapacity or substantial disruption of the ability to conduct normal life functions.
•
Results in a congenital anomaly/birth defect.
•
Is an important medical event that may not result in death, be life threatening, or require hospitalization, but based on appropriate medical judgment may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes listed above.
The term “severe” is used to describe the intensity of a specific AE. The AE itself may be of relatively minor clinical significance (such as a severe headache). Severe is not the same as “serious”.
Documentation of AEs
At every visit, the study staff will assess the occurrence of AEs. Reports of AEs between visits should be elicited by asking the subject a nonleading question such as “Do you feel different in any way since the last study infusion?”
AEs that occur during infusion or within 72 hours after infusion of test product, regardless of other factors that may impact a possible causal association with product administration, are defined as infusional AEs and are temporally associated with an infusion.
Infusional AEs (i.e., AEs temporally associated with an infusion of IVIG) may occur occasionally and include, but are not limited to, chills, headache, fever, vomiting, allergic reactions, nausea, arthralgia, low blood pressure and moderate low back pain. Rarely, IVIG may cause a sudden fall in blood pressure and, in isolated cases, anaphylactic shock, even when the subject has shown no hypersensitivity to previous administration. Cases of reversible aseptic meningitis, isolated cases of reversible hemolytic anemia/hemolysis and rare cases of transient cutaneous reactions have been observed with IVIG. Increases in serum creatinine and (or) acute renal failure have been observed. Very rarely, thromboembolic reactions such as myocardial infarction, stroke, pulmonary embolism, and deep vein thrombosis have been reported.
Certain severe adverse drug reactions may be related to the rate of infusion.
The recommended infusion rate must be closely followed. Subjects must be closely monitored and carefully observed for any symptoms throughout the infusion period.
Certain adverse reactions may occur more frequently at high infusion rates and in subjects with hypo- or agammaglobulinemia with or without IgA deficiency.
When subjects have been switched from another IVIG or when there has been an interval of 2 infusion cycles or more (e.g., ≥42 or 56 days) since the previous IVIG infusion, subjects should be monitored for 1 hour after the first infusion to detect potential adverse reactions. Other subjects should be observed for at least 20 min after administration.
For AEs that occur during infusion, (i) the infusion rate at the time of the onset of the AEs, (ii) the time of onset of AEs, and (iii) the time AEs change materially in intensity and (or) resolve will be recorded.
If an infusional AE occurs during the first treatment course, the study staff will initiate 1 of the following actions (graded from 1 to 4) depending on the nature and (or) severity of the event:
1.
Reduce the infusion rate to one-half the rate of the infusion at which the AE was observed; or
2.
Reduce the rate of the infusion more than one-half the rate at which the AE was observed, as necessary to subside symptoms; or
3.
Reduce the rate of the infusion to one-half, or progressively reduce to more than one-half the rate at which the AE was observed and then stop the infusion, as necessary to subside symptoms; or
4.
Stop the infusion to subside symptoms.
The study staff will evaluate the subject’s AE and then:
•
Will increase or resume the infusion at a rate tolerated by the subject once the symptoms have subsided; or
•
Will stop the infusion and not resume it.
The infusion rate schedule must be followed as listed in the section “Infusion Rates”.
If a subject has an infusional AE at the same infusion rate twice, then subsequent infusion escalations, if any, should be halted at the previously highest tolerated rate.
Assessment of AEs
Assessment of severity
The investigator will assess the severity of AEs according to the following criteria:
Mild: The AE is transient and does not interfere significantly with the subject’s normal functioning level. The AE resolves spontaneously or may require minimal therapeutic intervention.
Moderate: The AE produces limited functional impairment and may require therapeutic intervention. The AE produces no sequelae.
Severe: The AE results in significant impairment of function and may lead to temporary inability to resume the subject’s normal life pattern. The AE produces sequelae that require prolonged therapeutic intervention.
Assessment of causality
The following 4-point scale will be used by the investigator to rate the relationship of the adverse event to the study product:
Certain: The event follows a reasonable temporal sequence associated with the administration of the study product, follows a known or suspected response pattern to the product, is confirmed by improvement upon stopping the infusion and reappears upon repeated exposure (rechallenge).
Probable: The temporal relationship between the event and administration of the study product is suggestive, and the event is less likely to be explained by the subject’s medical condition or other therapies.
Possible: There is some temporal relationship between the event and the administration of the study product but the event could also be explained by the subject’s medical condition or other therapies.
Not related: An event for which insufficient information exists to conclude that the etiology of the event is unrelated to the investigational product.
The investigators will assign causality at their respective sites during the study. The medical monitor will review all AE and SAE data and other source documents to confirm the assigned causality for all AEs and SAEs prior to database lock. This assignment will be included in the study database and the final study report.
Reporting Serious Adverse Events (SAEs)
SAEs, regardless of the relationship to study medication or not, must be described in writing by completing a Serious Adverse Event form. The completed SAE form must be submitted to the sponsor/CRO via a designated 24/7 fax line.
Investigators should not wait to receive additional information to fully document the event before sending the SAE report.
The initial written notification must occur with 24 hours of learning about the SAE.
All initial, follow-up, and final SAE reports should be faxed to the sponsor/CRO.
Upon receipt, the sponsor/CRO will forward each SAE report to the medical monitor for review.
The medical monitor will be the primary contact for all SAE-related questions and concerns.
SAEs are to be followed until resolution or stabilization. Investigators must fax follow-up SAE forms to the medical monitor as additional information becomes available or as the status of the serious adverse experience changes. A final SAE form should be submitted once the event has resolved or stabilized.
Care should be taken by the investigator to record all concomitant medications taken by the subject in treatment of the event and, if hospitalized, to report all medications taken during hospitalization. Also, if hospitalized, all associated adverse experiences that occur during hospitalization should be reported on the case report form.
The sponsor will report in an IND safety report any suspected adverse reaction that is both serious and unexpected.
Data Safety Monitoring Board (DSMB)
An independent third-party DSMB will monitor the safety of the subjects on a periodic basis. Members of the DSMB will be independent of the study sponsor and participating sites. Safety parameters that will be evaluated by the DSMB and the sponsor will include:
•
Serious, including life-threatening or fatal, adverse drug experiences that are clearly related to the investigational product.
•
Other IND Safety Reports, CIOMS I and Health Canada’s Adverse Drug Reaction (ADR) which are submitted to the FDA according to 21CFR 312.32 requirements and the requirements of Health Canada, respectively.
•
Adverse drug reactions and suspected adverse drug reactions that are considered unexpected.
•
Suspected thrombotic events.
The DSMB may recommend, to the sponsor, that the trial be modified or terminated based on safety observations.
Warnings and precautions
Based on published clinical trials of IVIG products, the following AEs may be expected after treatment with the study IVIG:
•
Immediate reactions: mild reactions including fever, malaise, myalgia, and headache occurring during or within hours of completing the infusion have been reported in about 4% of patients. These symptoms often respond to a reduction in the infusion rate.
•
Inflammatory and allergic reactions: A transient rash, urticarial or macropapular, has been observed in up to 6% of patients. Eczema, erythema multiforme, purpuric erythema, and alopecia are also dermatological disorders that have been attributed to IVIG, but the mechanisms are unknown.
•
Thrombotic events: The symptoms include headache, fatigue, blurred vision, and thromboembolic complications. Patients with concomitant paraproteinemia or elderly patients with low cardiac output and (or) atherosclerotic stenosis may be prone to adverse effects from increased plasma viscosity.
•
Aseptic Meningitis Syndrome: Characterized by headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea, and vomiting is a rare AE whose exact mechanism is not clear. The frequency of this AE is not established. It is clustered mainly in patients with a history of migraine when high dosages (1 g/kg) are infused in short periods of time.
•
Hemolysis: Hemolytic anemia can develop subsequent to IVIG therapy due to enhanced RBC sequestration. IVIG recipients should be monitored for clinical signs and symptoms of hemolysis. Individuals infused with IVIG who develop positive direct antiglobulin (Coombs) should be monitored for hemolysis.
•
Transfusion-related lung injury (TRALI): Characterized by severe respiratory distress, pulmonary edema, hypoxemia, abnormal left ventricular function, and fever, and it typically occurs within 1–6 hours after infusion.
Statistical methods and sample size
Statistical design/method
The FDA has determined that trials of new IgG products can be open-label, single-arm trials compared with a statistically modeled historical control. The study should demonstrate that the upper one-sided 99% confidence limit of serious bacterial infections is ≤1.0 per person per year.
Moreover, the proportion of infusions for which 1 or more temporally associated AE has been reported should be have an upper one-sided 95% confidence limit of less than 0.40.
Sample size
A sufficient number of subjects should be studied to provide at least 80% power with one-sided hypothesis testing and an alpha = 0. FDA anticipates that studies of 40 to 50 subjects will be adequate (FDA/CBER 2008).
Analysis populations
The intention to treat (ITT) population will consist of all subjects who are enrolled in the study and who received any amount of study product.
The per-protocol (PP) population will consist of all subjects in the ITT population who complete the entire 12-month study period and who do not have any major protocol violations.
The pharmacokinetic population will be defined as a subset of the ITT population who participate the PK study.
The primary safety and efficacy analysis populations will be the same as the ITT population.
The PP population will also be analyzed for efficacy.
Demographics
Demographic data will be summarized overall by using the standard set of summary statistics, or by frequency tables, as appropriate. The standard set of summary statistics will consist of the mean, median, standard deviation, maximum, and minimum.
Efficacy
A formal statistical analysis (a noninferiority test) will be applied for the first primary efficacy endpoint. A 99% one-sided upper confidence limit for the incidence of serious acute bacterial infections will be derived, and conformance with the FDA standard will be considered acceptable if this limit is less than 1.0 per subject per year.
Secondary efficacy endpoints
The secondary endpoints will be analyzed descriptively. For hospitalizations, the number and (total) duration for each subject will be calculated and converted to a figure per year. These data will be presented using the standard set of summary statistics (except that the mean and standard deviation will be calculated weighting for the duration of data available for each subject).
Pharmacokinetic endpoints
Pharmacokinetic parameters will be estimated by noncompartmental analysis for each subject in the pharmacokinetics population. AUC will be determined using the (linear up/log down) trapezoidal rule. IgG pharmacokinetic parameters will be estimated. Secondary endpoints will be summarized descriptively. Where relevant, confidence intervals will also be presented. In the event of changes in dose or dosing interval, some adjustment of mean values (using data from the PK profile) may be required. Pharmacokinetic parameters will be summarized with descriptive statistics by infusion schedule and across all subjects.
The trough levels of Total IgG, IgG subclasses, and specific antibodies will also be summarized with descriptive statistics and confidence intervals.
Safety endpoints
To estimate the overall probability of the occurrence of an AE possibly related to infusion for study subjects, as well as a one-sided 95% upper confidence limit, the Generalized Estimating Equation (GEE) method of Zeger and Liang (1986) will be used.
AEs will be listed individually by body system with subject identification numbers and the overall incidences of all AEs that occur during or within 1 hour, 24 hours, and 72 hours following an infusion of test product, regardless of other factors that may impact a possible causal association with product administration, will be reported.
We will report the number of infusions administered, the total number of AEs reported at any time during the study (including AEs that the sponsor or the investigator determine were not product related).
For AEs that occur during infusion, we will report and analyze (i) the infusion rate in effect at the time of onset of adverse events, (ii) the time of onset of adverse events, and (iii) the time AEs change materially in intensity and (or) resolve. We will list SAEs, AEs by severity, AEs by body system, and our determination of which AEs were product related and which were not.
Viral safety data will be listed (not tabulated) and any seroconversions will be discussed individually in the text.
Level of significance
Confidence intervals will be calculated at the (2-sided) 95% level of confidence (except for the primary efficacy and safety endpoints which will be 1-sided 99% and 95%, respectively).
Statistical software
The statistical software to be used for analysis will be defined in the statistical analysis plan.
Ethics
Investigational Review Board/Research Ethics Board
International Conference of Harmonization (ICH) Topic E6 Good Clinical Practice (GCP) Guidelines (ICH/GCP) require that an Institutional Review Board (IRB) (Independent Ethics Committee or IEC in Canada) oversee all investigational drug studies. This board or committee, the makeup of which must conform to applicable regulations and guidelines, will approve all aspects of the study, including the protocol, written Informed Consent Form, and any subject information sheets to be used, prior to initiation of the study. The investigator will provide the sponsor with a copy of the communication from the committee to the investigator indicating approval of the protocol and consent form/information sheets. All amendments to the protocol must be reviewed and approved prior to implementation except where necessary to eliminate apparent immediate hazards to human subjects.
The investigator will be responsible for obtaining annual IRB/IEC renewal and submitting SAE reports to the IRB/IEC for the duration of the study (as per site policies and procedures). Copies of the investigator’s report and/or copies of the IRB/IRC extension approval must be sent to the sponsor.
Protocol deviations and violations will be submitted to IRB/IEC according to the requirements of each of these institutions.
Informed consent and assent
No subject can enter the study or the pharmacokinetic assessment before his/her informed consent has been obtained. The investigator will not undertake any investigation specifically required only for the clinical study until valid consent has been obtained. When a subject deemed legally incompetent, such as a minor child, is able to give assent to decisions about participation in research, the investigator must obtain that assent in addition to the consent of the legally authorized representative.
Subjects will be permitted to take the Informed Consent Form home and talk it over with others before they sign it. The consent (and, if applicable, the assent) process will be documented in study source records for each subject. The terms of the consent and the date and time of day when it was obtained must be documented in the CRF. The consent form, with the date and time of day when it was signed, must be retained by the Investigator as part of the study records. A copy of the signed Informed Consent Form will be given to the subject or subject’s legally authorized representative.
The ICF must be submitted by the investigator with the protocol to the IRB for approval.
Should a protocol amendment be required, the Informed Consent Form may be revised to reflect the changes of the protocol.
If the consent form is revised, it is the investigator’s responsibility to ensure that an amended consent form is reviewed and approved by the IRB or ethics committee and that this amended form is signed by all subjects subsequently entered in the study as well as those currently in the study.
Confidentiality
The investigator will ensure that the subjects’ anonymity will be maintained. As applicable, the privacy rules of the U.S. Health Insurance Portability and Accountability Act (HIPAA) and Canada’s Personal Health Information Protection Act, 2004, S.O. 2004, c.3 (PHIPA) will be followed to obtain authorization for most uses and disclosures of Protected Health Information (PHI). On CRFs or other documents submitted to the sponsor or its designee, subjects will not be identified by their names, but by an identification code, consisting of the combination of subject’s initials and study number. Documents not submitted to the sponsor (e.g., the site confidential subject enrollment log and original subjects’ consent forms), will be maintained by the investigator in strict confidence.
Declaration of Helsinki
The study will be conducted in accordance with the Declaration of Helsinki (1964) including all amendments up to and including the South Africa revision (1996).
Quality control and quality assurance
General information
This clinical trial will be conducted in accordance with the GCP/ICH guidelines. The sponsor or designee will systematically control the essential documents generated during this trial. All phases of the trial will be monitored by sponsor or designee with the critical phases of this trial, particularly the starting and the ending of the trial, being subject to internal audits by QA. All Clinical Study Monitoring visits and inspections by QA will be followed by internal reports and corrective actions, if needed. Follow-up letters will be forwarded to sites after all clinical visits.
Quality control by the monitoring team
The Clinical Study Monitors will monitor the data collected throughout the study. The investigator must be available for Clinical Study Monitors during their visits and must ensure that the Clinical Study Monitor has access to all documents that they require, including to the subject’s files (direct access).The investigator agrees to cooperate with the Clinical Study Monitor to make certain that any problems detected in the course of these monitoring visits are resolved. The anonymity of the subject must be safeguarded and all data checked during these monitoring visits must remain confidential.
Quality assurance by an audit team
Any study site may be selected for audit at any moment by an audit team originating from the sponsor or from an external organization acting on behalf of the sponsor. The investigator agrees to cooperate with the auditor to ensure that any problems detected in the course of these audit visits are resolved. The anonymity of the subject must be safeguarded and data checked during these monitoring visits must remain confidential.
Data handling and record keeping
Direct access to source data/documents
The investigator will permit study-related monitoring, audits, IRB review, and regulatory inspection(s), providing direct access to source data and documents.
Data collection and management
Data generated as per protocol will be entered onto the CRF in accordance with the parameters set forth in ICH Guideline for Good Clinical Practice (ICH Guideline For Good Clinical Practice E6(R1) 1996). When the CRFs have been completed, a monitor, with the assistance of the study site coordinator, will verify the source documentation records and review the data.
Any errors detected by either the clinical study monitor or the investigator after query resolution should be communicated via CRF data change forms.
Coding of AEs will be performed using the MedDRA dictionary. Similarly, coding of all medications will occur using the WHOMED dictionary. SAEs will be coded using MedDRA.
Record keeping
The investigator is responsible for maintaining all records pertaining to the clinical trial and for ensuring complete and accurate documentation.
The sponsor requires that each investigator retain records (all regulatory documents such as the protocol, study approval letters, all CRFs, drug dispensing and accountability logs, all original subject consent forms and all correspondence pertaining to the conduct of the study) for a period of no less than 2 years from the date of final regulatory approval or as per local regulations, whichever is longer.
Changes in the conduct of the study
The investigator and the sponsor must both agree to any change to this protocol prior to its implementation. Any protocol amendment must be submitted for information/consideration to the applicable Regulatory Agencies.
IRB/IEC approval will be requested for any change to this protocol which could affect the safety of subjects, the scope or design of the study, any increase in dosage, duration of exposure to study medication, an increase in the number of subjects treated by 10% or more, the addition of a new test or procedure, or the dropping of a test intended to monitor safety.
Reporting and publication
The sponsor or CRO will prepare a draft study report after the completion of the study. The final draft study report will be submitted to the investigators for information, review, and comments.
Publication of data generated in the study is governed by the Investigator Clinical Trial Agreement.
Liabilities and insurance
The study sponsor will pay for all study related costs. A separate financial agreement will be made (as appropriate) with the investigator and/or institutions.
In case of any damage or injury occurring to a subject in association with the trial medication or participation in the study, the sponsor will have an insurance policy.
Footnotes
1
Physical Exams may be performed by any physician listed on the FDA Form 1572 as a primary investigator or sub-investigator.
2
Blood chemistry includes total bilirubin, creatinine, BUN, alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, lactate dehydrogenase (LDH), glucose, sodium, potassium, chloride, CO2, and calcium.
3
Hematology panels include complete blood count with white blood cell differential and platelet count.
4
Urinalysis should include microscopic examination of urine sediment and pregnancy testing.
REFERENCES
Bergbreiter B. and Salzer U.2009. Common variable immunodeficiency: A multifaceted and puzzling disorder. Expert Rev. Clin. Immunol.5(2):167–180.
Bonilla F.A. and Geha R.S.2003. 12. Primary immunodeficiency diseases. J. Allergy Clin. Immunol.111(2 Suppl):S571–S581.
Eijkhout H.W., Der Meer J.W., Kallenberg C.G., Weening R.S, van Dissel J.T., Sanders LAM, Strengers P.F.W., Nienhuis H, and Schellekens P.T.A.2001. The effect of two different doses of intravenous immunoglobulin on the incidence of recurrent infections in subjects with primary hypogammaglobulinemia. A randomized, double-blind, multicenter crossover trial. Ann. Intern. Med.135:165–174.
FDA/CBER. 2008. Guidance for Industry: Safety, Efficacy, and Pharmacokinetic Studies to Support Marketing of Immune Globulin Intravenous (Human) as Replacement Therapy for Primary Humoral Immunodeficiency.
Pierce L.R. and Jain N.2003. Risks associated with the use of intravenous immunoglobulin. Transfus. Med. Rev.17(4):241–251.
ICH Guideline For Good Clinical Practice E6(R1). 1996.
International Union of Immunological Societies. 1999. Primary immunodeficiency diseases. Report of an IUIS Scientific Committee. Clin. Exp. Immunol.118(Suppl 1):1–28.
Roifman C.M. and Gelfand E.W.1988. Replacement therapy with high dose intravenous gamma-globulin improves chronic sinopulmonary disease in subjects with hypogammaglobulinemia. Pediatr. Infect. Dis. J.7:S92–S96.
Rosen F.S., Cooper M.D., and Wedgwood R.J.1995. The primary immunodeficiencies. N. Engl. J. Med.333(7):431–440.
Schroeder, H.W., Jr. Primary antibody deficiencies. In: Clinical Immunology Principles and Practice. Mosby International Ltd., 2001.
Yong P.F.K., Chee R., and Grimbacher B.2008. Hypogammaglobulinemia. Immunol. Allergy Clin. North Am.28:691–713.
Zeger S.L. and Liang K.Y.1986. Longitudinal data analysis for discrete and continuous outcomes. Biometrics.42:121–130.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 2 • June 2016
Pages: 67 - 85
History
Received: 15 February 2016
Accepted: 4 May 2016
Accepted manuscript online: 10 May 2016
Version of record online: 10 May 2016
Copyright
© 2016.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
John A.Hooper. 2016. A clinical trial protocol for the study of intravenous immunoglobulin in patients with primary antibody immunodeficiency disorders. LymphoSign Journal.
3(2): 67-85. https://doi.org/10.14785/lymphosign-2016-0005
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
