Abstracts from The Immunodeficiency Canada–CSACI symposium
Coronin-1A deficiency presenting with severe combined immunodeficiency
Adi Ovadiaa,b, Chaim M. Roifmana,b
aDivision of Clinical Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada; bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada
aDivision of Clinical Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada; bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada
Introduction: Coronins are a conserved family of actin-binding proteins that are important regulators of the actin cytoskeleton. They function in actin-dependent processes such as cytokinesis, cell motility, phagocytosis and vesicular trafficking. Coronin-1A is the predominant coronin expressed in lymphocytes. Mutations in CORO1A gene, encoding Coronin-1A, were described to cause variable degrees of T cell lymphopenia, susceptibility to infection and immune dysregulation.
The first reported patient presented with a severe combined immunodeficiency (SCID) phenotype due to a complete loss of the coronin-1A. This patient had a compound heterozygous mutations with one allele demonstrating interstitial deletion of chromosome 16p11.2, the region containing CORO1A, and the second allele with a 2 nucleotide frame-shifting deletion. Since then six additional patients of three kindred have been reported. Most of the patients presented with a clinical phenotype of Epstein–Barr Virus-associated lymphoproliferative disease. Some of the patients had no detectable Coronin-1A protein whereas three patients of one kindred had a hypomorphic mutation with a significantly decreased expression of the protein.
Three of the reported patients had a cognitive, developmental, and speech delay as well as behavioral abnormalities including attention deficit hyperactivity disorder (ADHD). The neurodevelopmental abnormalities were explained by chromosome 16p11.2 deletion in two of the patients and highly consanguineous family in the other patient.
Methods: We report a patient who presented in infancy with SCID phenotype and Bacillus Calmette-Guerin (BCG) lymphadenitis due to a missense mutation in CORO1A causing Coronin-1A deficiency and underwent successful hematopoietic stem cell transplantation (HSCT). The patient also demonstrated developmental delay and ADHD features.
Results: Our patient is a female born to a mother of Inuit descent following an unremarkable pregnancy. She received the BCG vaccine at birth. She presented at 6 months of age with a clinical history of recurrent chest infections starting at 2 months of age, followed by chronic suppurative otitis media, recurrent urinary tract infections with Escherichia coli and Klebsiella as well as chronic diarrhea, failure to thrive and oral thrush. At the age of 7 months, she was noticed to have an enlarged axillary lymph node. The lymph node was surgically excised and pathological analysis revealed Mycobacterium bovis, which is the same strain as the BCG vaccine. She was started on anti-mycobacterial treatment.
She started to demonstrate developmental delay at the age of 6 months with decreased axial and peripheral muscle tone.
Her initial immune evaluation revealed profound lymphopenia with low CD3+ T cells including both CD4+ and CD8+. She had normal CD19+ and NK cells as well as normal levels of immunoglobulins. Surprisingly, in vitro responses to phytohemaggutinin and anti-CD3 were comparable to control. The T cell receptor excision circles (TREC) level was very low. Circulating T cell repertoire analysis by flow cytometry revealed some underrepresented families (6,4,5,3,9,14,18) and others were overexpressed (2,3,7,1,21,3).
Genetic analysis by whole exome sequencing revealed homozygous mutation c.[602G>A] in CORO1A resulting in amino acid change from arginine to histidine (R201H).
The immunological manifestation of significant T cell deficiency led to the decision to preform HSCT. At the age of 16 months our patient underwent HSCT with peripheral blood stem cells from a 9/10 HLA-matched unrelated donor. She received myeloablative conditioning with busulfan and cyclophosphamide. Graft-versus-host disease (GvHD) prophylaxis consisted of alemtuzumab, cyclosporine and methylprednisolone. She continued to receive her anti-mycobacterial treatment. She had neutrophil engraftment on day +17, and had an unremarkable post-transplant course.
At 6 months post‐transplant, she developed acute GvHD of the skin and pancytopenia. She was treated with pulse doses of methylprednisolone with good response. She had an additional episode of hemolytic anemia a year later that was treated with a high dose of intravenous immunoglobulin (IVIg). One and a half years post-transplant she developed alopecia involving approximately 50% of the scalp which resolved with topical steroid treatment.
At 2 years post-transplant she achieved full immune reconstitution and we were able to discontinue her IVIg replacement therapy and the antimycobacterial medication.
Developmentally, at 4 years of age she still had expressive language delay and had inattentive, impulsive and hyperactive behavior diagnosed as ADHD.
Conclusion: This is the first report of a patient of Inuit descent presenting with SCID phenotype due to autosomal recessive mutation in CORO1A. As well, this is the first description of homozygous missense mutations in CORO1A causing Coronin-1A deficiency. Our patient underwent successful HSCT with full immune reconstitution and is currently doing well. Interestingly, she demonstrates developmental delay as well as features of ADHD, which suggests that the decreased coronin-1A expression is the cause for the neurodevelopmental abnormalities.
Progressive neurodegenerative manifestations in a patient with STAT1 dysfunction due to a novel mutation in the SH2 domain
Adi Ovadiaa,b, Nigel Sharfeb, Cynthia Hawkinsc, Suzanna Laughlind, Chaim Roifmana,b
aDivision of Clinical Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada; bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada; cDepartment of Laboratory Medicine & Pathobiology, The Hospital for Sick Children, Toronto, ON, Canada; dDepartment of Radiology, The Hospital for Sick Children, Toronto, ON, Canada
aDivision of Clinical Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada; bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada; cDepartment of Laboratory Medicine & Pathobiology, The Hospital for Sick Children, Toronto, ON, Canada; dDepartment of Radiology, The Hospital for Sick Children, Toronto, ON, Canada
Introduction: Signal transducers and activators of transcription (STATs) are a family of cytokine dependent latent DNA binding factors activated through tyrosine phosphorylation primarily by the Janus protein-tyrosine kinases (JAKs) as well as others. STAT1 serves as a cellular mediator and an essential effector of interferon α and interferon γ signaling as well as many other receptor signal pathways. The STAT1 structure contains an N-terminus, a coiled coil domain, DNA binding domain (DBD), a Src homology 2 (SH2) domain and a C-terminus domain that facilitates transcriptional induction.
Mutations in STAT1 have been described in a variety of clinical conditions and manifestations such as chronic mucocutaneous candidiasis (CMCC), autoimmunity and cancer. In rare cases STAT1 mutations have been associated with Moya-Moya like disease with recurrent cerebral vascular infracts due to vasculitis. Most of the mutations reported so far were heterozygous mutations located at the coiled coil domain proposed as gain of function mutations. Monoallelic mutations predominately in the DBD of STAT1 were also reported and are associated with a gradual decline in cellular and humoral immunity leading to fatal viral infections. Mutations in the SH2 domain of STAT1 were previously described as loss of function mutations demonstrating decreased STAT1 phosphorylation and resulted in Mycobacterial infections as a cause of Mycobacterial infections.
Method: We report of a patient with a novel mutation in the SH2 domain which results in gain of phosphorylation and leads to a unique CNS disorder.
Results: A 30‐year‐old female of English descent, born pre‐term at 32 weeks of gestation, suffered necrotizing enterocolitis soon after birth. She first presented at the age of 1 year with recurrent pneumococcal chest infections and otitis media. At the age of 13 years she was diagnosed with pneumatocele and left lower lobe bronchiectasis. Since bronchiectasis was progressive yet localized she underwent left lower lobe lobectomy with segmental resection of the left upper lobe at the age of 16 years with complete resolution of symptoms. She started to suffer from chronic oral and vaginal candidiasis at the age of 6 years and was diagnosed with primary ovarian failure at the age of 15 years. At the age of 17 years she started to complain of headaches and syncope and was found to have Arnold–Chiari malformation with secondary hydrocephalus, tonsillar herniation, and syringomyelia and underwent decompression surgery with complete resolution of symptoms.
At 26 years of age she suddenly developed difficulty in walking which progressed to right side weakness and spasticity as well as dysarthria. Investigations including visualized evoked potentials (VEPs), brain stem auditory evoked responses (BEARs) and somatosensory evoked potentials (SSEPs) were normal. CSF was negative for infectious agents and no oligoclonal banding was formed. Muscle biopsy showed no inflammation, necrosis or vascular changes, although concentric laminated bodies were observed in some of the fibers, which is a non-specific finding.
Evaluation of the immune system showed a normal IgG level of 12.6 g/L, IgA of 1.1 g/L and IgM 0.7 g/L. Lymphocyte markers demonstrated normal numbers of CD19(1012), CD3 (1903), CD4 (1012) and CD8(784) positive cells but slightly low NK cells at 348. Specific antibody levels were protective to Rubella, Varicella, and Tetanus. Proliferative responses to mitogens were normal but in-vitro response to antigens such as Candida, CMV, simplex, and zoster were absent.
Analysis of the STAT1 gene revealed heterozygous mutation +/c. 1885C>T in the SH2 domain predicting a histidine to tyrosine amino acid change at position 629 (H629Y).
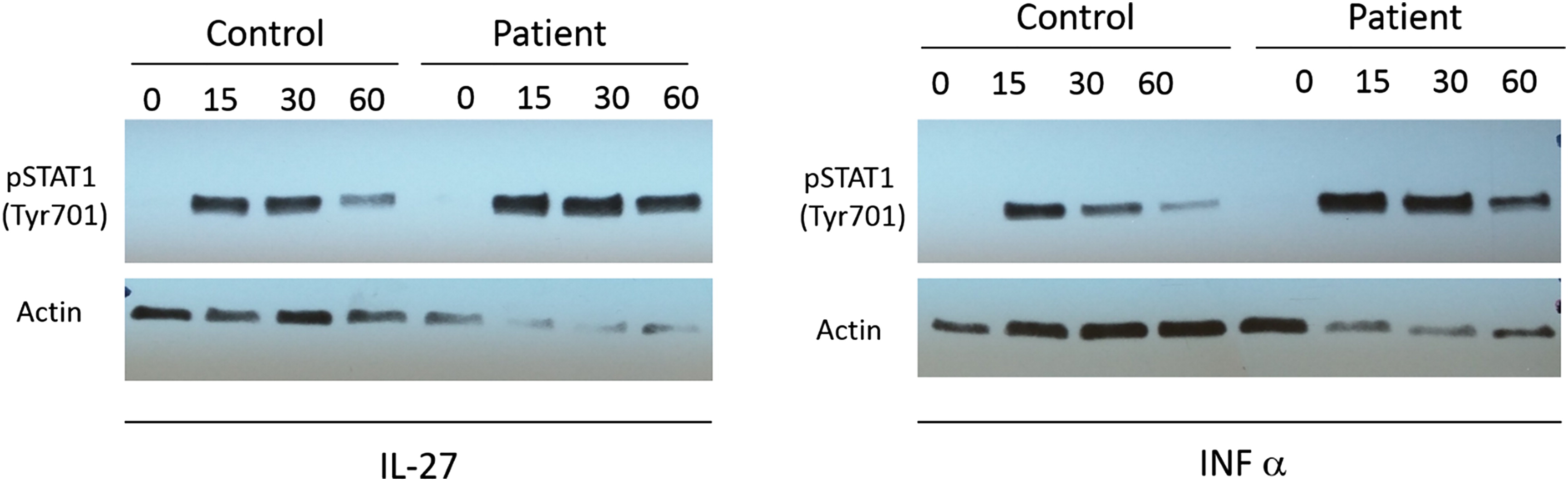
The activation of STAT1 was evaluated by western blot analysis of phosphorylated STAT1 in mature activated T cells following stimulation with IFN-α and IL-27. Our patient demonstrated increased level of STAT1 activation by pY701 STAT1 compared to a healthy control (Figure 1). Interestingly, when we preformed analysis of the STAT1 expression in the same T cells, there was a clear increase in STAT1 expression in our patient compared with control. This difference was also demonstrated when we measured the expression of STAT1 in PBMCs and cultured T cells.
Figure 1:
MRI of the brain and the spinal cord demonstrated diffuse abnormalities in the periventricular region and in the white matter with extensive wellerian degeneration in the brainstem affecting mainly the pyramidal tract. CT angiogram of the brain was unremarkable.
Brain biopsy revealed evidence of vasculopathy demonstrated by marked thickening and hyalization of the small vessels wall predominantly in the white matter.
Immunostaining with NOTCH-3 specific monoclonal antibody demonstrated sparse granular labeling of vascular smooth muscle cells and some small vessels in the cortex and white matter. NOTCH-3 is expressed only in vascular smooth muscle cells in human adult tissues and its immunostaining is used as a marker for the neurodegenerative disease such as Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL). The sparse labeling of NOTCH3 in our patient is similar in distribution but less in intensity to CADASIL suggestive of NOTCH3 pathway abnormality.
Treatment with monthly intravenous pulse methylprednisolone was initiated as the neurological symptoms continued to progress. At first it seemed to be alleviating the patient's symptoms but repeat doses did not seem to have additional effect, and therefore treatment was discontinued after six months. Soon after she was started with monthly high dose of IVIG, again with some temporary minor gain. Because her disease continued to progress we escalated therapy with mycophenolate mofetil (MMF). This treatment seems to attenuate the progression of the neurological deterioration with minor gains in motor skills.
Conclusion: We describe a new neurodegenerative manifestation associated with STAT1 mutation. The CNS disorder is unique as it is not a result of vasculitis, aneurism, or recurrent cerebral vascular ischemia, but most probably is secondary to an inflammatory process. This is the first report of heterozygous STAT1 SH2 domain mutation with gain of phosphorylation presenting with early onset of airway bacterial infections and significant autoimmunity.
Progressive ataxia and recurrent pneumonia in a 20-month-old male
Alexander Lyttlea, Anne K. Junkera,b, Stuart E. Turveya,b, Kyla J. Hildebranda,b
aDivision of Allergy and Clinical Immunology, Department of Pediatrics, British Columbia Children's Hospital, Vancouver, BC, Canada; bChild & Family Research Institute, University of British Columbia, Vancouver, BC, Canada
aDivision of Allergy and Clinical Immunology, Department of Pediatrics, British Columbia Children's Hospital, Vancouver, BC, Canada; bChild & Family Research Institute, University of British Columbia, Vancouver, BC, Canada
Case Presentation: A term 20 month-old male of Russian-Mennonite descent from rural British Columbia was urgently assessed at a tertiary care pediatric Immunology center for a history of recurrent pneumonia, worsening asthma symptoms and progressive ataxia. A chronic productive cough began at 2 months of age followed by prolonged and recurrent upper respiratory tract infections treated with intermittent inhaled corticosteroids and 7 courses of oral prednisone for presumed viral-induced asthma. Referral was made to a tertiary care center asthma specialist at 15 months and daily asthma inhaler therapy with high dose mometasone/formoterol was recommended with no improvement in cough symptoms. Further history documented five pneumonias (confirmed with chest x-ray), three of which required admission to a rural hospital for oxygen supplementation and once required intravenous (IV) antibiotics. Parents reported 8 episodes of acute otitis media. At 15 months, loose and frequent stools were noted lasting four weeks with recurrence at 19 months. Diaper dermatitis was intermittently noted with poor response to topical treatments. There were no other invasive, unusual, or recurrent infections. He began walking at 12 months of age; however, at 13 months he began falling with ambulation which progressively worsened with age. Immunizations had been administered and tolerated including live vaccines. Parents declined the rotavirus vaccine. The family history was positive for breast cancer in the paternal great-grandmother and one paternal aunt. Both grandmothers had a history of two spontaneous abortions. Parents and patient's siblings (sisters, ages 2 and 8 years; brother, 10 years) were healthy. The family acknowledged no consanguinity; however, the parents lived in the same small Mennonite community. Physical examination demonstrated hypertelorism, marked truncal ataxia and difficulty standing length 89 cm, weight 11 kg, and head circumference 45.5 cm (95th, 25th, and <3rd percentiles, respectively). Ocular and cutaneous telangiectasias were present bilaterally on the bulbar conjunctiva and overlying the maxillae. Oral thrush was noted, lymph nodes were palpable and tonsillar tissue was present. Respiratory exam found increased work of breathing, cough, diffuse crackles, and wheeze on auscultation. Excoriated diaper dermatitis consistent with Candida was noted. The remainder of examination was unremarkable.
Lymphopenia (0.7 cells × 109 cells/L) and hypogammaglobulinemia (IgG 1.51 g/L) were noted as early as 9 months of age and persisted. At 15 months, markedly decreased absolute T lymphocyte counts (CD3 0.28 cells× 109 cells/L) and B lymphocytes (0.18 cells × 109 cells/L) were detected; IgA, IgM levels, and NK cell levels were normal; and vaccine titres were indeterminate to measles and rubella, however protective for tetanus (1.28 IU/mL) and diphtheria (1.16 IU/mL). On admission to hospital, mitogen stimulation found poor responses to Pokeweed antigen, Concanavalin A, and Staphylococcus aureus. Urinary purines and pyrimidines were negative. Stool studies were positive for Candida and negative for viruses; 8 AM serum cortisol level was 248 nmol/L (240–618). Bronchoalveolar lavage (BAL) fluid was positive for respiratory syncytial virus, Enterococcus cloacae, and Streptococcus pneumoniae. A diagnosis of ataxia telangiectasia (A-T) was made based on clinical and laboratory phenotype, and this diagnosis was further reinforced by an elevated alpha fetal protein level of 66 µg/L (normal <8). Shortly after the BAL procedure the patient developed tachypnea and hypotension requiring 60 cc/kg of fluid boluses and attributed to likely septic shock. He received a two-week course of IV antibiotics and oral fluconazole was given for ongoing diarrhea secondary to Candida infection. Monthly IV immunoglobulin replacement was initiated (600 mg/kg/dose) as well as antibiotic prophylaxis with trimethoprim-sulfamethoxazole and amoxicillin (as a bridge to obtaining therapeutic trough IgG levels). The inhaled corticosteroids were discontinued. The patient remains infection-free following hospital discharge. The father has subsequently learned that he has a cousin with two children likely affected by A-T, one of which died at age 8 years from a malignancy. Genetic test results are pending.
Discussion: A-T is an autosomal recessive neuro-degenerative disorder caused by a mutation in the ataxia-telangiectasia mutated (ATM) gene located on chromosome 11. A-T has a prevalence of 1 in 20 000–100 000 (Swift et al. 1986) but has been reported in higher frequency in populations such as Amish and Mennonites and a founder mutation is known in this patient's community (Telatar et al. 1998).
ATM is a kinase responsible for the phosphorylation of many proteins, including p53, a tumour suppressor gene involved in stopping the cell cycle between the G1 and S phase. Without proper p53 function, the cell is unable to make repairs to double stranded DNA breaks such as those seen in V(D)J recombination and leads to higher prevalence of malignancy, radiation sensitivity and combined immunodeficiency (Suarez et al. 2015). A prospective study assessing 100 consecutive patients with A-T found 71% of A-T patients had some form of immunodeficiency (Nowak-Wegrzyn et al. 2004). All of these patients were lymphopenic to varying degrees with CD4 T-cells decreased in 69%, CD8 T-cells in 51% and CD19+ B cells in 75%. Many patients had dysgammaglobulinemia with the most common antibody deficiencies impacting levels of IgG4 (65%), IgA (65%), IgG2 (48%), IgE (23%), and IgG (18%). In vitro lymphoproliferation assays to antigens and mitogens were decreased in 42% and 35%, respectively. Common infections included recurrent sinopulmonary infections and cutaneous warts. Less commonly varicella, herpes and molluscum contagiosum were observed. Two studies have shown the immunodeficiency in A-T is non-progressive (Nowak-Wegrzyn et al. 2004; Chopra et al. 2013).
Clinical Immunology and Allergy specialists frequently assess patients referred for asthma or recurrent viral wheezing. This case reinforces key clinical features that should prompt consideration for primary immunodeficiency including: asthma symptoms unresponsive to high-dose therapy, recurrent sinopulmonary infections, persistent lymphopenia, and neurological developmental regression. Further dissemination of tools such as the Jeffery Modell Foundation 10 Warning Signs for Immunodeficiency may lead to earlier recognition of these conditions and referral to pediatric immunology centers. At a subspecialist level, this case also demonstrates the need for subspecialty practitioners to remain connected with families who have lost children to rare immune disorders. In this case, heightened family and community awareness of the risk for A-T could have promoted carrier detection, breast cancer screening with magnetic resonance imaging rather than mammography, and earlier diagnosis and appropriate management of affected children.
REFERENCES
Chopra, C., Davies, G., Taylor, M., Anderson, M., Bainbridge, S., Tighe, P., and McDermott, E.M. 2013. Immune deficiency in Ataxia-Telangiectasia: A longitudinal study of 44 patients. Clin. Exp. Immunol. 176: 275–282. PMID: 24387201. doi:https://doi.org/10.1111/cei.12262.
Nowak-Wegrzyn, A., Crawford, T.O., Winkelstein, J.A., Carson, K.A., and Lederman, H.M. 2004. Immunodeficiency and Infections in Ataxia-Telangiectasia. J. Pediatr. 144: 505–511. PMID: 15069401. doi:https://doi.org/10.1016/j.jpeds.2003.12.046.
Suarez, F., Mahlaoui, N., Canioni, D., Andriamanga, C., Dubois d'Enghien, C., Brousse, N., Jais, J.-P, Fischer, A., Hermine, O., and Stoppa-Lyonnet, D. 2015. Incidence, presentation, and prognosis of malignancies in ataxia-telangiectasia: A report from the French national registry of primary immune deficiencies. J. Clin. Oncol. 33(2): 202–208. PMID: 25488969. doi:https://doi.org/10.1200/JCO.2014.56.5101.
Swift, M., Morrell, D., Cromartie, E., Chamberlin, A.R., Skolnick, M.H., and Bishop, D.T. 1986. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am. J. Hum. Genet. 39(5): 573. PMID: 3788973.
Telatar, M., Teraoka, S., Wang, Z., Chun, H.H., Liang, T., Castellvi-Bel, S., Udar, N., Borresen-Dale, A.L., Chessa, L., Bernatowska-Matuszkiewicz, E., Porras, O., Watanabe, M., Junker, A., Concannon, P., and Gatti, R.A. 1998. Ataxia-telangiectasia: Identification and detection of founder-effect mutations in the ATM gene in ethnic populations. Am. J. Hum. Genet. 62(1): 86–97. PMID: 9443866. doi:https://doi.org/10.1086/301673.
A case of primary intestinal lymphangectasia presenting as severe combined immunodeficiency
Bahar Torabi, Bruce D. Mazer
Introduction: Primary intestinal lymphangectasia is a disorder with an unknown underlying etiology mostly affecting children in the first three years of life. It is characterized by dilated and tortuous lymphatics in the small bowel mucosa and submucosa resulting in protein-losing enteropathy. It can present with diarrhea, hypoalbuminemia, malnutrition, and edema. It can also mimic an immunodeficiency syndrome with lymphopenia and hypogammaglobulinemia. This case describes a male infant with lymphopenia, hypogammaglobulinemia, and chronic diarrhea referred to our Immunology clinic for assessment of severe combined immunodeficiency (SCID).
Case Presentation: A term 7‐month‐old boy of Algerian descent was initially seen in our Immunology clinic with lymphopenia, hypogammaglobulinemia, and chronic diarrhea. He was well from birth until 2 months of age when he developed diarrhea with up to 10 watery stools per day after receiving the rotavirus vaccine. He was exclusively breastfed initially, but due to the persistent diarrhea, he was switched to a hydrolyzed formula, then to an amino acid-based formula, with some improvement of his diarrhea.
His investigations revealed a white blood cell count of 4.9 × 103 cells/mm3 and a lymphocyte count of 1.0 × 103 cells/mm3. The total protein was 30 g/L, albumin 17 g/L, with undetectable IgG and IgA. He had markedly low vaccine responses to tetanus, diphtheria, and Hemophilus influenza, with significant T cell lymphopenia (CD3 0.9 × 103 cells/mm3), and poor lymphocyte proliferation. His urinalysis and sweat chloride test were normal. A chest X-ray and echocardiogram were unremarkable.
He was immediately started on Pneumocystic jirovecii and antifungal prophylaxis, as well as immunoglobulin replacement. With time, the lymphocyte proliferation improved and the hypogammaglobulinemia resolved on immunoglobulin therapy; however, he remained hypoalbuminemic and lymphopenic. Gastrointestinal biopsies revealed focal lymphangectasia and nodular lymphoid hyperplasia. He was started on medium chain triglycerides orally with subsequent improvement of his hypoalbuminemia and lymphopenia. Once the lymphocyte count and lymphocyte proliferation normalized, his prophylactic treatments were stopped. He has been off immunoglobulin replacement for the past year with normal levels and vaccine serologies.
Conclusion: Secondary causes of immunodeficiency, such as primary intestinal lymphangectasia, should be suspected even in young infants. In this case of lymphangectasia, with protein losing enteropathy, the salient immune findings included lymphopenia and hypogammaglobulinemia. An unexpected finding was severely decreased lymphocyte proliferation, which returned to normal with improved nutrition and correction of his protein and albumin levels.
CD40 deficiency: more than a class switch recombination and somatic hypermutation defect. A review and report of an adult patient with a particular hyper immunoglobulin M syndrome
Luis Murguia-Favelaa,b, Nigel Sharfeb, Ariana Karanxhab, Harjit Dadib, Andrea Batesb, Chaim M. Roifmana,b
aDivision of Clinical Immunology and Allergy, Department of Paediatrics; bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, the Hospital for Sick Children and the University of Toronto, Toronto, ON, Canada
aDivision of Clinical Immunology and Allergy, Department of Paediatrics; bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, the Hospital for Sick Children and the University of Toronto, Toronto, ON, Canada
Background: CD40 deficiency is a rare autosomal recessive combined primary immunodeficiency characterized by defects of immunoglobulin class switch recombination (CSR) and somatic hypermutation (SMH) that is part of an expanding group of diseases collectively known as Hyper immunoglobulin M syndromes (Davies and Thrasher 2010). This defect not only affects immune cells. Signaling through CD40 is important to endothelial cells and neurons, among others (Karmann et al. 1995; Tan et al. 2002; Fontana et al. 2003).
CD40/CD40 Ligand interaction in B and T cells occurs after antigen-mediated activation and it is an absolute requirement to initiate CSR and SHM by promoting germinal center development in secondary lymphoid organs, the location where these processes take place. This cross-linking induces germinal centers to proliferate and to be suitable for CSR and SHM but also protects B cells from undergoing apoptosis and generates long-lived plasma cells (Notarangelo et al. 2006, 2007).
Following cross linking of CD40 on the B cell membrane, and given that this molecule lacks kinase activity on its cytoplasmic domain, intracellular signaling happens through the activation of TNF receptor associated factors (TRAFs), particularly TRAF-2, -3, -5, and -6. These factors function as adaptor molecules to recruit different kinases (e.g., mitogen-activated protein kinases or MAPKs) resulting in activation of transcription factors such as NF-κB (Vogel and Noelle 1998; Lougaris et al. 2005).
Clinical manifestations of the disease usually begin early in life with recurrent sinopulmonary bacterial infections and susceptibility to opportunistic organisms. Management of CD40 deficient patients consists of aggressive antibiotic treatment of infections and immunoglobulin replacement therapy (Lougaris et al. 2005). In contrast with CD40 Ligand deficiency, where the defect resides mainly on T cells, treatment with hematopoietic stem cell transplantation (HSCT) for CD40 deficiency is theoretically not a complete cure. It has been attempted in four patients with successful immune reconstitution in three (Kutukculer et al. 2003; Mazzolari et al. 2007; Al-Dhekri et al. 2012).
Since its description in 2001 (Ferrari et al. 2001) only fourteen patients with CD40 deficiency from ten unrelated families have been reported to date, and with the exception of one patient of Italian descent, the rest are from Middle Eastern countries (Kutukculer et al. 2003; Mazzolari et al. 2007; Al-Dhekri et al. 2012; Al-Saud et al. 2013). We report an additional adult patient of Iranian/French Canadian decent with a novel mutation in CD40, presenting later in life and with a milder phenotype. Also, in contrast with the patients reported previously, our patient's mutation allows CD40 expression in the cell membrane and adds 37 amino acids to the cytoplasmic domain of the protein, which seems to affect one of the two known TRAF2 binding sites linked to the MAP-kinases pathway while the NF-κB pathway appears to activate normally (preliminary data).
Methods: Clinical data were collected by retrospective chart review and directly from the patient. Informed consent was obtained. Genomic DNA was extracted from whole blood samples obtained from the patient. Promoters, exons, and both the 5’ and 3’ untranslated regions of CD40 were amplified by polymerase chain reaction for forward and reverse sequencing using the previously designed primers. RNA was also extracted from his Epstein–Barr virus (EBV)-transformed B cell line. CD40 expression on B cells was assessed by flow cytometry. Western blot analyses were performed following standard protocols. The complete manuscript, currently in preparation, will describe methods in more detail.
Results: Clinical Features. The patient is a now a 20 year-old man born to non-consanguineous parents. His father is from Iran and his mother is French-Canadian. The patient was reported to have had motor developmental delay during his first 2 years of life and aversion to foods of unclear etiology requiring gastrostomy tube feedings until he was 3 years of age. His development improved afterwards. He first presented to us at the age of 15 years with a history of three documented pneumonias and one episode of septic arthritis, all presenting after he was 10 years of age. He had a history of frequent acute otitis media between the ages of 1 and 4 years that resolved afterwards without the need of tympanostomy tubes or other measures. He was healthy until the age of 10 years when he had the first episode of pneumonia. The second and third pneumonias occurred at the ages of 12 and 13 years, respectively. Also at the age of 13 years he experienced an episode of right knee septic arthritis for which we do not know the causative organism. He recovered fully from all of these infections after completing adequate antibiotic courses.
Immunological features. When he was assessed at the age of 15 years he was found to have undetectable (<0.1 g/L) titers for both IgG and IgA (normal ranges of 4.5–14.3 and 0.2–1.0 g/L, respectively) and elevated IgM of 19.2 g/L (0.2–1.8 g/L). He did not have protective titers of specific antibodies for the vaccines that he had received despite a complete immunization schedule. He had normal numbers of B cells and T cells (CD19+ 454, CD3+ 2526, CD4+ 1670, CD8+ 721, NK cells 161) and his lymphocyte proliferation assays to both mitogens and antigens were also normal. He was then commenced on monthly intravenous immunoglobulin replacement therapy and has continued until now without any other episodes of severe or recurrent infections. As there was never any evidence of opportunistic infections he was not started on prophylactic antibiotics. He has remained clinically well.
Mutation analysis. Molecular analysis showed normal CD40 Ligand and AID gene sequences. Analysis of CD40 gene revealed a novel homozygous 1 base pair deletion in Exon 9 of both genomic and complementary DNA. The mutation c.823delG results in p.E275RfsX38 that translates into a frame shift that adds 37 extra amino acids into the protein.
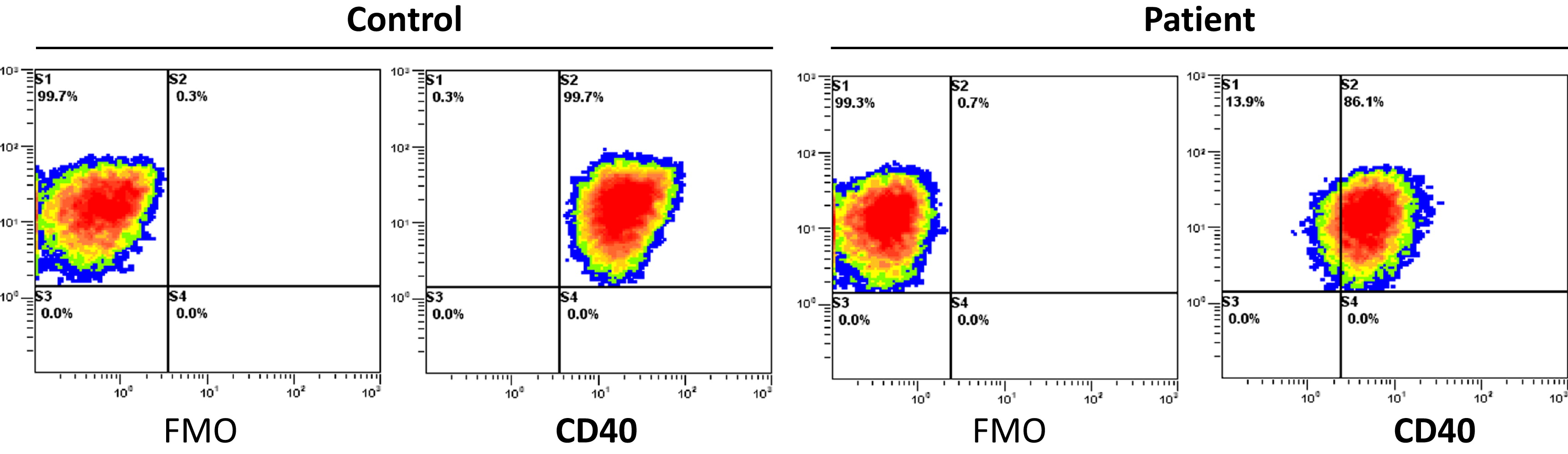
CD40 expression. CD40 is expressed in the surface of the patient's EBV-transformed B cell line as shown by Figure 1.
Figure 1:
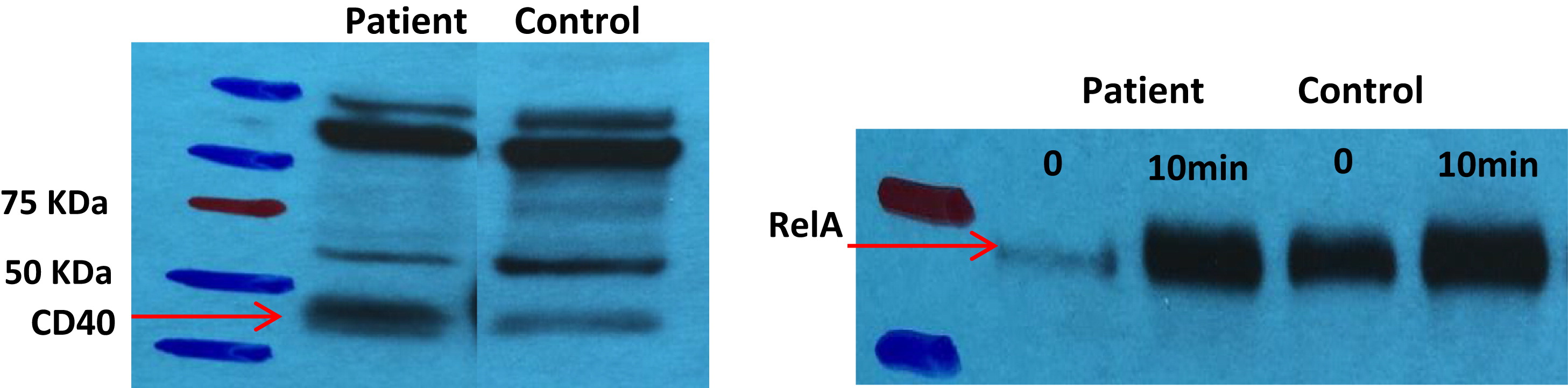
Western blot analyses. Expression of CD40 protein in our patient's EBV-transformed B cells was also confirmed by Western blot analysis. Preliminary results show normal activation of the NF-κB pathway (Figure 2).
Figure 2:
Conclusions: We report an adult patient with CD40 deficiency that presented to us at a later age and manifested with a milder phenotype as compared with previously reported patients. Also, in contrast with these patients, our patient has a novel mutation that allows expression of CD40 molecule in the surface of his B cells and adds 37 amino acids to the intracellular domain of this receptor likely affecting one of the two known TRAF-2 binding sites linked to the MAP-kinases signaling pathway ultimately resulting in the CSR and SMH defects. Until fully understanding the action of CD40 in lymphocytes and other cell types, we must remain sensible to identify all the possible phenotypes, and thus the importance of reporting our patient and his particularities as we learn more from the disease.
REFERENCES
Al-Dhekri, H., Al-Sum, Z., Al-Saud, B., Al-Mousa, H., Ayas, M., Al-Muhsen, S., Arnaout, R., Al-Seraihy, A., Hawwari, A., and Al-Ghonaium, A. Successful outcome in two patients with CD40 deficiency treated with allogeneic HCST. Clin. Immunol. 143(1): 96–98. PMID: 22342113. doi:https://doi.org/10.1016/j.clim.2012.01.012.
Al-Saud, B.K., Al-Sum, Z., Alassiri, H., Al-Ghonaium, A., Al-Muhsen, S., Al-Dhekri, H., Arnaout, R., Alsmadi, O., Borrero, E., Abu-Staiteh, A., Rawas, F., Al-Mousa, H., and Hawwari, A. 2013. Clinical, immunological, and molecular characterization of hyper-IgM syndrome due to CD40 deficiency in eleven patients. J. Clin. Immunol. 33(8): 1325–1335. PMID: 24122029. doi:https://doi.org/10.1007/s10875-013-9951-9.
Davies, E.G., and Thrasher, A.J. 2010. Update on the hyper immunoglobulin M syndromes. Br. J. Haematol. 149(2): 167–180. PMID: 20180797. doi:https://doi.org/10.1111/j.1365-2141.2010.08077.x.
Ferrari, S., Giliani, S., Insalaco, A., Al-Ghonaium, A., Soresina, A.R., Loubser, M., Avanzini, M.A., Marconi, M., Badolato, R., Ugazio, A.G., Levy, Y., Catalan, N., Durandy, A., Tbakhi, A., Notarangelo, L.D., and Plebani, A. 2001. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc. Natl. Acad. Sci. U.S.A. 98(22): 12614–12619. PMID: 11675497. doi:https://doi.org/10.1073/pnas.221456898.
Fontana, S., Moratto, D., Mangal, S., De Francesco, M., Vermi, W., Ferrari, S., Facchetti, F., Kutukculer, N., Fiorini, C., Duse, M., Das, P.K., Notarangelo, L.D., Plebani, A., and Badolato, R. 2003. Functional defects of dendritic cells in patients with CD40 deficiency. Blood. 102(12): 4099–4106. PMID: 12893749. doi:https://doi.org/10.1182/blood-2003-04-1244.
Karmann, K., Hughes, C.C., Schechner, J., Fanslow, W.C., and Pober, J.S. 1995. CD40 on human endothelial cells: Inducibility by cytokines and functional regulation of adhesion molecule expression. Proc. Natl. Acad. Sci. U.S.A. 92(10): 4342–4346. PMID: 7538666.
Kutukculer, N., Aksoylar, S., Kansoy, S., and Cetingul, N., and Notarangelo, L.D. 2003. Outcome of hematopoietic stem cell transplantation in hyper-IgM syndrome caused by CD40 deficiency. J. Pediatr. 143(1): 141–142. PMID: 12915844. doi:https://doi.org/10.1016/S0022-3476(03)00274-9.
Kutukculer, N., Moratto, D., Aydinok, Y., Lougaris, V., Aksoylar, S., Plebani, A., Genel, F., Notarangelo, L.D. 2003. Disseminated cryptosporidium infection in an infant with hyper-IgM syndrome caused by CD40 deficiency. J. Pediatr. 142(2): 194–196. PMID: 12584544. doi:https://doi.org/10.1067/mpd.2003.41.
Notarangelo, L.D., Lanzi, G., Peron, S., and Durandy, A. 2006. Defects of class-switch recombination. J. Allergy Clin. Immunol. 117(4): 855–864. PMID: 16630945. doi:https://doi.org/10.1016/j.jaci.2006.01.043.
Notarangelo, L.D., Lanzi, G., Toniati, P., and Giliani, S. 2007. Immunodeficiencies due to defects of class-switch recombination. Immunol. Res. 38(1–3): 68–77. PMID: 17917012. doi:https://doi.org/10.1007/s12026-007-0023-1.
Lougaris, V., Badolato, R., Ferrari, S., and Plebani, A. 2005. Hyper immunoglobulin M syndrome due to CD40 deficiency: Clinical, molecular, and immunological features. Immunol. Rev. 203: 48–66. PMID: 15661021. doi:https://doi.org/10.1111/j.0105-2896.2005.00229.x.
Mazzolari, E., Lanzi, G., Forino, C., Lanfranchi, A., Aksu, G., Ozturk, C., Giliani, S., Notarangelo, L.D., and Kutukculer, N. 2007. First report of successful stem cell transplantation in a child with CD40 deficiency. Bone Marrow Transplant. 40(3): 279–281. PMID: 17502893. doi:https://doi.org/10.1038/sj.bmt.1705713.
Tan, J., Town, T., Mori, T., Obregon, D., Wu, Y., DelleDonne, A., Rojiani, A., Crawford, F., Flavell, R.A., and Mullan, M. 2002. CD40 is expressed and functional on neuronal cells. EMBO J. 21(4): 643–652. PMID: 11847112. doi:https://doi.org/10.1093/emboj/21.4.643.
Vogel, L.A., and Noelle, R.J. 1998. CD40 and its crucial role as a member of the TNFR family. Semin. Immunol. 10(6): 435–442. PMID: 9826576.
Ataxia Telangiectasia detected on Ontario newborn TREC screening: first impression counts most
Mariam Hannaa, Vy Kimb, Brenda Reidb, Peter B. Denta, Chaim M. Roifmanb,c,d
aDivision of Immunology/Rheumatology, McMaster University, Hamilton, ON, Canada; bDivision of Immunology/Allergy, Department of Pediatrics, Hospital for Sick Children, Toronto, ON, Canada; cDepartment of Immunology, University of Toronto, Toronto, ON, Canada; dCanadian Centre for Primary Immunodeficiency, The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, Toronto, ON, Canada
aDivision of Immunology/Rheumatology, McMaster University, Hamilton, ON, Canada; bDivision of Immunology/Allergy, Department of Pediatrics, Hospital for Sick Children, Toronto, ON, Canada; cDepartment of Immunology, University of Toronto, Toronto, ON, Canada; dCanadian Centre for Primary Immunodeficiency, The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, Toronto, ON, Canada
Introduction: Newborn Screening (NBS) has recently included screening programs aimed at detection of severe combined immunodeficiency (SCID) through enumeration of T-cell receptor excision circles (TRECs). While screening has detected infants with SCID, other combined immunodeficiencies have also been reported through this screening test. We report the first case of ataxia telangiectasia (AT) detected by NBS in Canada.
Case Description: A term male infant was born in Ontario to non-consanguineous Caucasian parents. The pregnancy history and family history were non-contributory. He had NBS via blood spot analysis completed on Day 2 of life as per protocol. TREC screening returned a positive result of 41 copies/3 µL (normal 83 – 3316/3 µL). The Ontario cutoff had recently been adjusted to 75 copies/3 µL. Deletion analysis of the TBX1 (22q11 critical region) did not detect a deletion with 2 normal copies. Purine panel measured by tandem mass spectrometry was within normal limits; thus, screening was negative for adenosine deaminase and purine nucleoside phosphorylase deficiencies. Complete blood count done on Day 1 of life demonstrated lymphopenia (0.73 × 109/L). A repeat dried blood spot on Day 14 was surprisingly normal with 152 copies/3 µL and negative for TBX1 deletion, with a normal purine profile similar to previous. The patient continued to have lymphopenia (1.9 × 109/L). A second fresh whole blood sample on Day 14 demonstrated normal TREC level of 1227/0.5 µg of DNA (normal >400/0.5 µg of DNA). The patient was referred to a tertiary level center (Hospital for Sick Children, Toronto) for further assessment. At 3 months of age the whole blood TREC level was again reduced at 280/0.5 µg of DNA, and remained persistently low at 229/0.5 µg of DNA at 5 months of age. He continued to have persistent lymphopenia (1.22 × 109/L). Lymphocyte immunophenotyping by flow cytometry demonstrated B- and T- cell lymphopenia (CD19+ 154 × 106/L, CD4+ 421 × 106/L, CD8+ 224 × 106/L). Mitogen stimulation was completed at 3, 4, and 5 months of age and revealed an initially borderline response to phytohaemagglutinin of less than 50% of control, which subsequently normalized at 3 months. However, there was no response to anti-CD3 at 3, 4, or 5 months. At 5 months of age, the patient was noted to have immunoglobulin (Ig) G < 1.10 g/L, IgA < 0.10 g/L and IgM 0.60 g/L. Of note, the serum IgM level was relatively elevated compared to the IgG level. Anti-tetanus toxoid antibody was unprotective despite the patient having received 2 doses of tetanus vaccine. As such, intravenous Ig replacement therapy and prophylactic antibiotics were initiated. The infant has remained infection free at this time. His growth and development have been within normal limits to date. There is no clinical evidence of telangiectasias at this point in time. Whole exome sequencing detected 2 mutations in the ATM gene, consistent with a diagnosis of AT.
Discussion: This is the first case of AT detected by newborn screening in Canada. The detection limit of 75 copies/3 µL of TRECs allowed for this case to be detected. However, the subsequent normalizing TREC can be confusing for the clinician. The persistent lymphopenia, undetectable IgG and relatively elevated IgM levels are indicators of an immune defect in this patient. Earlier detection and diagnosis of AT in this patient allowed for intervention prior to the onset of infections, discussion of clinical prognosis, early referral for problems associated with this condition and genetic counseling. As well, the diagnosis of ATM prevented unnecessary procedures, such as hematopoietic stem cell transplantation and avoidance of ionizing radiation.
IRAK-4 deficiency as a cause for familial fatal invasive infection by Steptococcus pneumoniae
Victoria E. Cooka, Serge Graziolib, Sara J. Hamiltonc, Margaret L. McKinnonc, Kate L. Del Bela, Linda Hoangd, Kyla J. Hildebranda, Anne K. Junkera, Stuart E. Turveya
aDivision of Allergy and Clinical Immunology, Department of Pediatrics, British Columbia Children's Hospital and Child & Family Research Institute, University of British Columbia, Vancouver, BC, Canada; bPediatric Intensive Care Unit, Department of Pediatrics, British Columbia Children's Hospital, University of British Columbia, Vancouver, BC, Canada; cDepartment of Medical Genetics, Child & Family Research Institute, University of British Columbia, Vancouver, BC, Canada; dBC Centre for Disease Control Public Health Microbiology & Reference Laboratory, Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada
aDivision of Allergy and Clinical Immunology, Department of Pediatrics, British Columbia Children's Hospital and Child & Family Research Institute, University of British Columbia, Vancouver, BC, Canada; bPediatric Intensive Care Unit, Department of Pediatrics, British Columbia Children's Hospital, University of British Columbia, Vancouver, BC, Canada; cDepartment of Medical Genetics, Child & Family Research Institute, University of British Columbia, Vancouver, BC, Canada; dBC Centre for Disease Control Public Health Microbiology & Reference Laboratory, Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada
Here we report the case of two siblings with fatal pneumococcal meningitis as the initial manifestation of IRAK-4 deficiency caused by previously undescribed mutations in IRAK4. This letter highlights the importance of invasive pneumococcal infection as a critical “red flag” warning for potential underlying primary immunodeficiency, and identifies challenges in making the clinical diagnosis of IRAK-4 deficiency.
Pneumococcal meningitis in an adult with IRAK4 deficiency.
Vy H.D. Kima, Brenda Reida, Valerie Salesb, Chaim M. Roifmana,c,d
aDivision of Immunology and Allergy, Department of Pediatrics, Hospital for Sick Children, Toronto, ON, Canada; bDivision of Infectious Diseases, Department of Medicine, University Health Network and Markham Stouffville Hospital, Toronto, ON, Canada; cDepartment of Immunology, University of Toronto, Toronto, ON, Canada; dCanadian Centre for Primary Immunodeficiency, the Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, Toronto, ON, Canada
aDivision of Immunology and Allergy, Department of Pediatrics, Hospital for Sick Children, Toronto, ON, Canada; bDivision of Infectious Diseases, Department of Medicine, University Health Network and Markham Stouffville Hospital, Toronto, ON, Canada; cDepartment of Immunology, University of Toronto, Toronto, ON, Canada; dCanadian Centre for Primary Immunodeficiency, the Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, Toronto, ON, Canada
Background: Individuals with mutations in IL-1 receptor-associated kinase 4 (IRAK4) present in infancy and early childhood with severe invasive bacterial infections. Most patients have their first invasive infection before 2 years of age. The microbial pathogens most frequently identified in these patients include Staphylococcus aureus and Streptococcus pneumoniae. Curiously, a large cohort demonstrated that these patients stopped experiencing invasive infections during adolescence. We report here a patient who developed pneumococcal meningitis at the age of 32 years.
Methods: Consent was obtained from the patient for inclusion in the Canadian Centre for Primary Immunodeficiency (CCPID) Registry and tissue bank, which has been approved by the SickKids Research Ethics Board (protocol no. 1000005598). Data from medical records were compiled prospectively and retrospectively and entered into the CCPID Registry.
Case Description: The patient is one of three siblings who all carry a homozygous C to T transition at nucleotide 877 within exon 8 of the IRAK4 gene. This mutation creates a premature stop codon at amino acid 293 (Q293X). Both parents were heterozygous for this mutation. His sister died at 5 months of age due to fulminant S. aureus meningitis. His twin brother suffered multiple invasive infections, including S. pneumoniae meningitis at 2 years of age and Pseudomonas aeruginosa necrotizing epiglottitis at 5 years of age.
The patient had meningitis at the age of 10 months, septic arthritis at 2 years and bilateral tonsillar abscesses at 6 years. At the age of 8 years he had a brain abscess that presented with hemiparesis and dysarthria but with normal body temperature. At the age of 14 years he suffered from multiple abdominal abscesses. In all episodes but one, blood or aspiration cultures grew S. pneumoniae. At the age of 15 years he had an infection with Mycobacterium avium, which was detected in blood cultures.
This patient's detailed immune work up has been previously described elsewhere. In short, he had somewhat reduced T cell numbers but normal in-vitro responses to mitogens. His T-cell receptor excision circle level was normal. Immunoglobulin (Ig) G, IgM and IgA levels were normal as were responses to vaccinations with tetanus, polio, measles, mumps, and rubella. Responses to polysaccharide pneumococcal vaccine were variable with good responses to some but not to other serotypes. Because of the partial response to pneumococcal vaccine, he was kept on prophylaxis with trimethoprim-sulfamethoxazole since the age of 4 years. However, this did not prevent his invasive infections. Intravenous immunoglobulin (IVIg) was added to the prophylaxis regimen with presumably improved protection. We observed that as long as he received both modalities of treatment he was free of infection. At the age of 18 years he decided to discontinue the IVIg infusions and as well as prophylaxis with trimethoprim-sulfamethoxazole. He had no significant infections for a period of at least ten years.
His recent episode of meningitis occurred at the age of 32 years. It started with otitis media, treated with amoxicillin-clavulanate. He developed vertigo and headaches but seemed to have improved on this antibiotic regimen. Two weeks later he was seen again in the emergency department for fever and increased confusion. He received ceftriaxone and vancomycin and had a lumbar puncture. Acyclovir was added pending HSV/VZV PCR. He subsequently developed seizures and needed to be sedated and intubated in the intensive care unit. Analysis of his cerebral spinal fluid (CSF) showed 385 × 106/L WBC with 97% neutrophils and 3% lymphocytes. CSF glucose was less than 0.7 mmol/L and total protein was elevated at 4 g/L. CSF and blood cultures grew penicillin-resistant S. pneumonia sensitive to vancomycin and intermediately sensitive to ceftriaxone. Rifampin was added for synergy for part of the treatment. Computerized tomography scan of the brain showed encephalomalacic changes in the right frontoparietal region, unchanged from previous exams, right otomastoiditis with mastoid superior ridge bone loss, and diffuse chronic sinusitis. He gradually improved on antibiotic treatment allowing his discharge from hospital after two weeks with no major complications except right ear deafness. He has since received vaccination with conjugated followed by booster polysaccharide pneumococcal vaccines, quadrivalent conjugated meningococcal and typhoid vaccines.
Conclusions: This case should alarm physicians to the possibility of life threatening infections occurring beyond adolescence in patients with IRAK4 deficiency. As such, these patients should be counselled about the ongoing risk of infections and be aggressively monitored and treated, including for minor infections. Consideration should be made for ongoing preventative therapy with Ig replacement therapy, optimal vaccination schedule (if not on Ig replacement therapy), and prophylactic antibiotics, if indicated.
Hematopoietic stem cell transplantation for a patient with heterozygous STAT1 mutation
Vy H.D. Kima, Brenda Reida, Julia Uptona, Adelle R. Atkinsona, Eyal Grunebauma,b, Chaim M. Roifmana,b,c
aDivision of Immunology/Allergy, Department of Pediatrics, Hospital for Sick Children, Toronto, ON, Canada; bDepartment of Immunology, University of Toronto, Toronto, ON, Canada; cCanadian Centre for Primary Immunodeficiency, The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, Toronto, ON, Canada
aDivision of Immunology/Allergy, Department of Pediatrics, Hospital for Sick Children, Toronto, ON, Canada; bDepartment of Immunology, University of Toronto, Toronto, ON, Canada; cCanadian Centre for Primary Immunodeficiency, The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, Toronto, ON, Canada
Background: Patients with mutations in signal transducer and activator of transcription 1 (STAT1) manifest a range of clinical presentations including chronic mucocutaneous candidiasis, autoimmunity, and susceptibility to mycobacterial, fungal or viral infections. Recently, a group of patients with de novo heterozygous mutations in STAT1 have been described who had a form of progressive combined immunodeficiency with life-threatening severe infections and autoimmune features. These patients were identified as having mutations in or near the DNA binding domain of STAT1. The majority of these patients succumbed to overwhelming infection by the second decade of life. Hematopoietic stem cell transplantation (HSCT) has been reported in few patients with STAT1 mutations, however, none have survived. We report a case of a young patient with STAT1 mutation who underwent a successful HSCT.
Methods: Consent was obtained from the patient's parents for inclusion in the Canadian Centre for Primary Immunodeficiency (CCPID) Registry and tissue bank, which has been approved by the Sick Kids Research Ethics Board (protocol no. 1000005598). Data from medical records were compiled prospectively and retrospectively and entered into the CCPID Registry.
Case Description: The patient is a young male who was referred to the Immunology clinic at Sick Kids at the age of 2 years 10 months for recurrent fungal infections. He was the product of an uneventful pregnancy. His parents are nonconsanguineous and are of Caucasian descent. The family history was non-contributory. From the age of 20 months, he had recurrent infections of the skin and oral mucosa that were positive for Candida albicans. He had several episodes of otitis media and one episode of pneumonia that were treated with oral antibiotics as an outpatient. He had no endocrinopathy or gastrointestinal symptoms. His initial immunological evaluation demonstrated normal numbers of B- and T-lymphocytes, normal adenosine deaminase activity, and normal in vitro mitogen stimulation to phytohaemagglutinin (PHA). Over time, his T-lymphocyte counts and in vitro mitogen responses to PHA and anti-CD3 declined. Sanger sequencing of STAT1 revealed a de novo heterozygous mutation, T385M, in the DNA binding domain. This same mutation had previously been identified posthumously in 2 patients who died in the first decade of life from invasive infection.
At 6.5 years of age the patient received an allogeneic HSCT from his brother, who was a 10/10 HLA match. His brother was healthy and did not have any identifiable mutation in STAT1. The patient received myeloablative conditioning with busulfan (16 mg/kg) and cyclophosphamide (200 mg/kg). He started cyclosporine A (target levels 150–200 µg/L) on Day -3 and methylprednisolone (2 mg/kg) on Day 0 for graft versus host disease (GvHD) prophylaxis. He received regular intravenous immunoglobulin replacement therapy as bacterial prophylaxis and pentamidine for Pneumocystis jiroveci pneumonia prophylaxis.
On Day +6 post-HSCT, the patient developed grade 1 acute GvHD of the skin. On Day +20, he developed stage 2 acute GvHD of the gut with profuse diarrhea. He was treated with pulse doses of methylprednisolone (30 mg/kg/day × 3 days) and oral budesonide was added to the cyclosporine A. On Day +27 he developed frankly bloody stools requiring red blood cell transfusion. Endoscopies and gut biopsy at the time were consistent with acute GvHD of the gut and mycophenolate mofetil (target levels >0.14 mg/L) was added. It was weaned off quickly within 2 weeks and replaced with sirolimus (target levels 5–15 µg/L) due to the potential for mycophenolate mofetil to exacerbate gastrointestinal symptoms. On Day +45 the patient had a significant gastrointestinal bleed requiring multiple red blood cell and platelet transfusions. A technetium 99m labeled RBC study and computerized tomography study were consistent with GvHD and active bleeding from a branch of the superior mesenteric artery that was 12–15 cm proximal to the ileocecal valve with bowel wall thickening. Attempts at embolization were unsuccessful. Despite treatment with octreotide and proton pump inhibitors and restarting mycophenolate mofetil and another pulse of methylprednisolone, the patient continued to have significant lower gastrointestinal bleeding. He underwent resection of 25 cm of distal ileum and ileostomy placement on Day +74. Pathology of the resected ileum demonstrated blunted villi with focal mucosal flattening, increased numbers of crypt apoptosis, edematous and focally hemorrhagic lamina propria, and markedly edematous submucosa with focal acute hemorrhage. There were areas of ulceration with adjacent thrombi within small and medium-sized submucosal vessels. The pathology did not demonstrate any evidence for CMV, adenovirus, or EBV infection.
The patient's course was also complicated by Haemophilus influenza pneumonia on Day +31; a lung infection with Aspergillus fumigatus at 3 months post-HSCT that was treated with a prolonged course of amphotericin B; BK virus in the urine; liver dysfunction and renal impairment.
Over the course of his admission, the patient gradually showed improvement in acute GvHD of the skin and gut. Cyclosporine A was discontinued by 6 months post-HSCT and the patient was discharged from hospital at 10 months post-HSCT. Sirolimus, corticosteroids and mycophenolate mofetil were slowly weaned and discontinued at 11, 14, and 18 months, respectively. At 20 months post-HSCT, the patient is clinically well and continues to have full donor engraftment with >95% donor chimerism.
Conclusions: This patient is the first reported case of survival more than 1.5 years after HSCT in a patient with a heterozygous mutation in the DNA binding domain of STAT1. Although he received an HSCT from a matched sibling donor, he suffered a complicated post-transplant course with significant acute GvHD of the skin and gut, considerable lower gastrointestinal bleeding requiring ileal resection and fungal lung infection. The vascular involvement in the ileum may have been pre-existing and related to his underlying immunodeficiency even though he did not have gastrointestinal symptoms prior to the HSCT, as patients with STAT1 mutations have previously been described with autoimmunity and vascular abnormalities. Despite his complicated course, the patient is clinically well with full donor engraftment at more than 1.5 years post-HSCT. As patients with mutations in the DNA binding domain have susceptibility to severe infections and early mortality, hematopoietic stem cell transplantation could be considered for these patients.
Hemophagocytic lymphohistocytosis in a patient with CD3δ deficiency
Mohammed Alsalamaha, Amrita Sarpalb, Victoria Mok Siub,c, Paul Gibsonb, CA Ruparb,c,d, Michelle Barton, Marina I Salvadorib, Sharan Goobieb,c, Chaim Roifmana,d
aDivision of Immunology and Allergy, The Canadian Center for Primary Immunodeficiency, The Hospital for Sick Children, Toronto, ON, Canada; bDepartment of Pediatrics, London Health Sciences Centre, Departments of Pathology and Laboratory Medicine, University of Western Ontario, London, ON, Canada; cMedical Genetics Program of Southwestern Ontario; dThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, Toronto, ON, Canada
aDivision of Immunology and Allergy, The Canadian Center for Primary Immunodeficiency, The Hospital for Sick Children, Toronto, ON, Canada; bDepartment of Pediatrics, London Health Sciences Centre, Departments of Pathology and Laboratory Medicine, University of Western Ontario, London, ON, Canada; cMedical Genetics Program of Southwestern Ontario; dThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, Toronto, ON, Canada
This full version of this Original Article appears on page 201 of this issue (https://doi.org/10.14785/lpsn-2015-0006).
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 4 • December 2015
Pages: 213 - 226
History
Version of record online: 13 November 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
2015. Abstracts from The Immunodeficiency Canada–CSACI symposium. LymphoSign Journal.
2(4): 213-226. https://doi.org/10.14785/lpsn-2015-0904
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
