The effects of STAT1 dysfunction on the gut
Abstract
Introduction: Mutations in the signal transducer and activator of transcription1 (STAT1) have been associated with a variety of clinical patterns. Interestingly patients with heterozygous mutations in the DNA binding domain (DBD) of STAT1 suffer acute and chronic colitis.
Methods: To further analyze the role of STAT1 deficiency in intestinal inflammation, we employed protein expression analysis of total and activated STAT1 in intestinal biopsy samples from 2 patients with heterozygous mutations in the DBD of the STAT1 gene.
Results: Both patients showed clinical and histological features of colitis. Total and activated STAT1 were decreased in duodenal and colonic enterocytes, and total STAT1 was found to be mislocalized in aggregates subapically. In addition, intestinal biopsy samples showed decreased numbers of lymphocytes. Patient-derived lymphoblasts demonstrated lack of viability and high susceptibility for cell death.
Conclusion: STAT1 expression and distribution in the gut of patients with mutations in the DBD are abnormal, suggesting a primary role of STAT1 dysfunction in enterocytes in addition to the secondary effect of aberrant inflammation.
Statement of novelty: Colitis associated with STAT1 mutations appears to have unique features distinct from typical inflammatory bowel disease.
Introduction
The transcription factor Signal Transducer and Activator of Transcription 1 (STAT1) is playing a key role in controlling cellular responses to a variety of growth factors such as IL-2, IL-3, IL-6, IL-9, IL-10, IL-11, IL-12, IL-15, IFNα, IFNβ, and IFNγ as well as hormones such as the growth hormone.
Cytoplasmic STAT1 is activated by JAK kinases through tyrosine phosphorylation after phosphorylated STAT1 dimers translocate to the nucleus and bind to cognate target DNA sequences (Najjar and Fagard 2010; O'Shea et al. 2013).
Mutations in the STAT1 gene have been known for a while to be associated with susceptibility to mycobacterial and fungal infections. Several distinct groups have been identified so far. A fatal form of complete deficiency of STAT1 expression (Dupuis et al. 2001, 2003) renders patients susceptible to overwhelming viral or mycobacterial infections. Some heterozygous mutations predispose to weakly pathogenic mycobacteria (Chapgier et al. 2006, 2009). More recently, heterozygous mutations predominantly in the coiled-coil domain were found in cases with autosomal dominant chronic mucocutaneous candidiasis (Liu et al. 2011; van de Veerdonk et al. 2011; Maródi et al. 2012).
Increased STAT1 tyrosine phosphorylation and DNA binding, identified predominantly in EBV transformed lyphoblastic cell lines, derived from these patients led to their designation as gain-of-function mutations. Whether this increased phosphorylation truly leads to gain of function or dysfunction remains to be determined, especially in primary cells.
In contrast to this phenotypic group, heterozygous mutations predominantly in the DNA binding domain (DBD) appear to lead to a different and more severe phenotype at an earlier age. These mutations in patients develop over time, usually within the first decade of life; patients present with a combined immunodeficiency, they suffer multiple autoimmune manifestations, and they die young of viral infections or HLH (Takezaki et al. 2012; Sharfe et al. 2014; Faitelson et al. 2014; Roifman 2014).
Interestingly, these patients suffer acute and chronic colitis that may begin in infancy (Uzel et al. 2013; Sharfe et al. 2014). A detailed pathological and immunohistological assessment of the gut in these patients was first documented by Sharfe et al. (2014). However, it remained unclear whether this manifestation is caused by infection, autoimmunity, a primary gut epithelium defect, or a combination of these factors. Here we attempted to study primary changes in gut enterocytes as well as to assess gut inflammation.
Methods
Patient enrollment
Studies on the tissue material of patient 2 and the control samples were carried out with the approval of the Research Ethics Board at the Hospital for Sick Children, Toronto, Ontario. Therefore, an extensive and comprehensive consent process was used for both the patients and the control individuals. Informed consent to participate in the research including genetic analysis was obtained through the Canadian Center for Primary Immunodeficiency Registry and Tissue Bank (Protocol No. 1000005598). Consent for gut samples was obtained by the Pediatric IBD cohort study (NEOPICS), available on their website at http://www.neopics.org/NEOPICS Documents.html. Controls included (i) patients that had undergone duodenal and colonic biopsies for evaluation of gastrointestinal symptoms but whose endoscopy and biopsy reports appeared normal and (ii) patients with active Crohn’s Diesease and intestinal inflammation, but with a normal STAT1 gene.
Clinical features of patients
Patient 1 was previously described by Sharfe et al. (2014). Patient 2 was a 13-year-old female who was born prematurely at 34 weeks gestation and was treated for breathing difficulties at birth. Parents were unrelated and reported no history of immunodeficiency, autoimmunity, or cancer in their families. The patient suffered repeated episodes of bacterial pneumonias from the age of 12 months. After the use of nebulized steroids or antibiotics, she occasionally developed oral thrush and a perianal candida infection.
At 7 years of age she began complaining of intermittent abdominal pain and nausea. These episodes of pain were frequently associated with loose stools. Subsequently she gradually lost weight and reported chronic fatigue. Although her height continued to follow the 75th percentile, her weight declined to 25th percentile.
Lower gastointestinal endoscopy performed at 8 years of age showed multiple aphthous ulcers through the rectum, sigmoid, and ascending and transverse colon. Lymphoid hyperplasia was detected in the terminal ileum.
At the age of 13 years she continued to have acute exacerbations of chronic lung disease as well as chronic diarrhea and abdominal pain. An endoscopic examination revealed chronic active colitis with focal areas of acute cyrptitis and pericryptic and occasional crypt abscesses.
Immune evaluation showed normal lymphocyte counts with normal distribution of T-cell subpopulations. Immunoglobulin levels as well as specific antibodies remained mostly protective.
Genetic analysis
Genetic analysis of the patients, as well as the mutation of patient 1, was reported previously (Sharfe et al. 2014).
Patient 2 had a heterozygous +/V389A, c.1166C>T De novo mutation in the DBD of the STAT1 gene.
Histology analysis
Paraffin-embedded tissue sections were deparaffinized using Xylene, and they were then dehydrated with different percentages of ethanol. Slides were then stained for 5 min with hematoxylin and 5 min with eosin (Thermo Scientific). Next the stained tissue sections were mounted with Entellan mounting medium (EMD-Millipore). For periodic acid-Schiff cyto-chemistry, sections were incubated in 0.5% periodic acid solution (Sigma Aldrich) for 15 min at room temperature. Afterwards, the tissue was washed twice for 2 min with deionized water and incubated for 5 min in Schiff's reagent solution (Sigma Aldrich) in darkness. Tissue sections were then washed for 5 min at room temperature and counterstained with hematoxylin solution (Thermo Scientific). Finally, slides were mounted with Entellan. Photomicrographs were taken using a Leica Inverted Light Microscope and adapted for brightness, contrast, and pixel size using Adobe Photoshop CS5 Version 12.0.
Immunohistochemistry for T-lymphocyte markers CD3, CD4, and CD8
Immunohistochemistry stainings for CD3, CD4, and CD8 were performed with an automated immonostainer from Ventana (BenchMark XT), and they were previously described in detail by Ngan et al. (2014).
Multilabel immunofluorescence and confocal microscopy
Duodenal and colonic biopsies from controls and patients were fixed with 10% formaldehyde without methanol and embedded in paraffin. Paraffin from the embedded tissue sections was removed using Xylene, and then it was rehydrated with different percentages of ethanol. An antigen retrieval step was performed with high-pressure cooking with 1 mM EDTA at a pH of 9 with 0.05% Tween 20. Slides were then blocked for 1h at room temperature with 5% BSA in 1× phosphate-buffered saline (PBS, MultiCell) without Ca and Mg containing 15% goat serum. Primary antibody incubation was performed overnight at 4°C. On the following day, stained slides were washed 3 times for 10 min with 1× PBS without Ca and Mg. Secondary antibody incubation was performed at room temperature and darkness for 1 hr, and slides were then washed 3 times for 10 min in darkness. Next, nuclear counterstaining with Hoechst 33342 Fluorescence Stain (Thermo Scientific) was performed at a dilution of 1:15 000. Finally, sections were mounted overnight with Vectorshield fluorescence mounting medium (Vector Labs).
Rabbit-monoclonal STAT1 (Cell Signalling), rabbit-monoclonal phospho-STAT1 (Y701, Cell Signalling), and mouse-monoclonal antibeta catenin (BD Transduction Laboratories) were used as primary antibodies in a dilution of 1:100. ALEXA 568 goat antirabbit (Life Technologies) and ALEXA 488 goat-antimouse (Life Technologies) served as secondary antibodies in a dilution of 1:500. Photomicrographs of immunostained sections were taken with an Olympus IX81 inverted fluorescence microscope equipped with a Hamamatsu C9100-13 back-thinned EM-CCD camera and Yokogawa CSU X1 spinning disk confocal scan head. Images were adjusted for contrast and brightness using the Volocity 6.1.1 version software (Perkin Elmer).
Lymphoblast culture, cell growth, and viability analysis
EBV transformed lymphoblasts from both patients and controls were and cultured in RPMI containing 10% fetal bovine serum (Gibco) and 1% Pen/Strep (Gibco). To assess cell growth and viability, lymphoblasts were seeded in a total number of 200 000 cells per each sample. Cells were analyzed for number and viability on day 2, day 4, and day 6. Cell counting and viability assessment were performed with the Countess™ Automated Cell Counter (Invitrogen). Cell growth and viability was measured in duplicates of 2 independent experiments. Statistical significance was calculated using the unpaired student t test. The final p value was p ≤ 0.05, meaning all values under or equal to 0.05 were assumed as being statistically significant.
Protein expression analysis
For protein expression analysis of total and activated STAT1, cells were harvested and lysed in lysate buffer (50 mM Tris HCl, 200 mM NaCl, 50 mM NaF, 1 mM EDTA, 20 mM beta-Glycerophosphat, and 1% Triton X-100), and then they were loaded on a gradient SDS page gel (4%–20%, Biorad). Rabbit-monoclonal STAT1 (Cell Signalling), rabbit-monoclonal phospho-STAT1 (Y701, Cell Signalling), mouse-monoclonal alpha Tubulin (Sigma Aldrich), and mouse-monoclonal GAPDH (Thermo Scientific) served as first antibodies in a dilution of 1:1000. Secondary HRP-conjugated antirabbit and antimouse antibodies (Sigma Aldrich) were used at a dilution of 1:2000 for antimouse and 1:5000 for antirabbit antibodies.
Results
Histopathology analysis of patients with mutations in STAT1
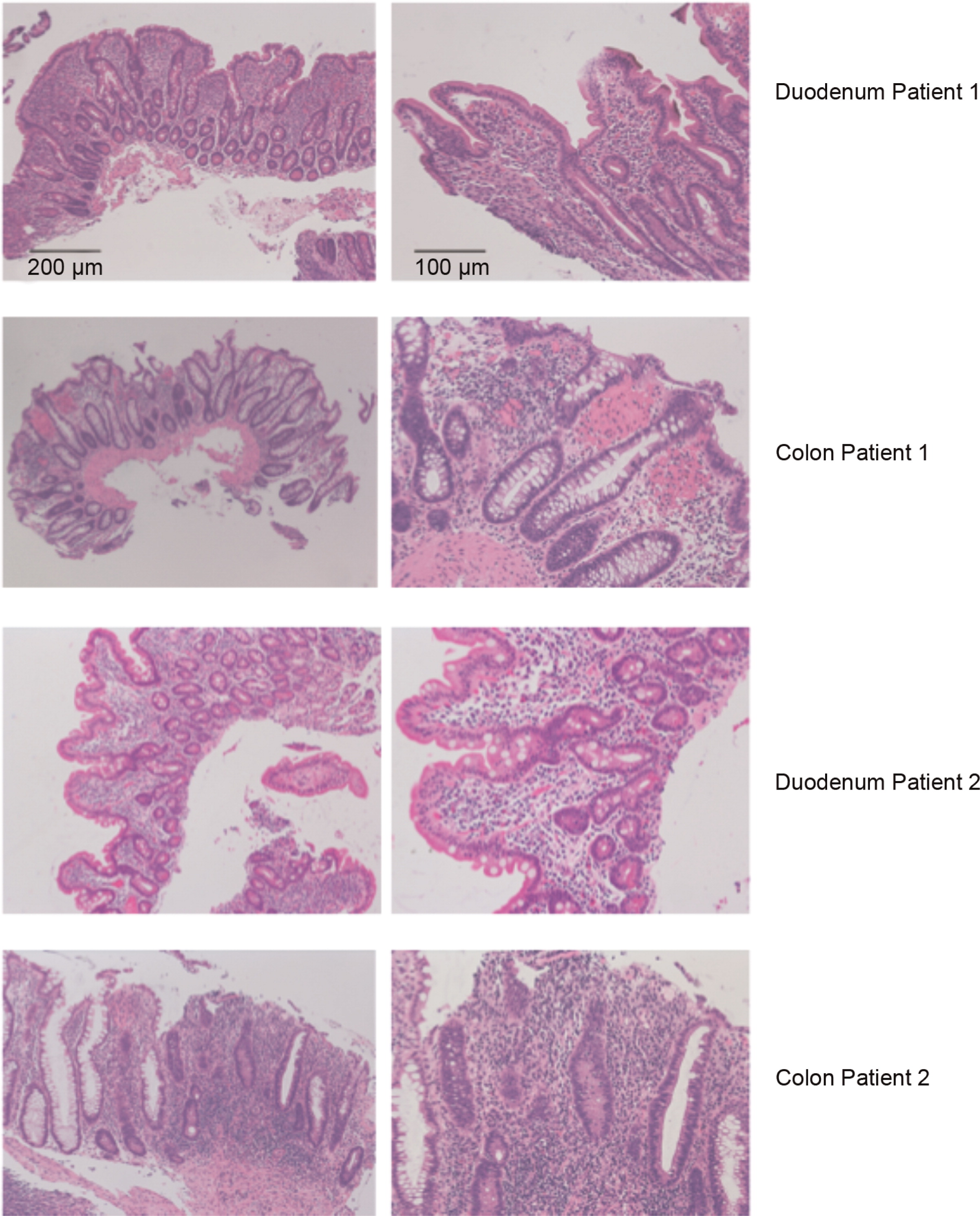
Both patients presented with chronic intestinal symptoms requiring upper and lower endoscopy and biopsy. Hematoxylin and eosin staining of the patients’ biopsy samples demonstrated features consistent with intestine inflammation (Figure 1). Patient 1 showed duodenal villous atrophy and villous blunting as well as increased infiltration of interstitial inflammatory cells, consistent with acute duodenitis (Figure 1). Colonic biopsy samples of patient 1 showed focal colitis with areas of increased interstitial inflammatory cells (Figure 1). Similarly, patient 2 showed focal inflammation within the colon (Figure 1).
Figure 1:
Lymphocyte populations in patients’ intestinal biopsies
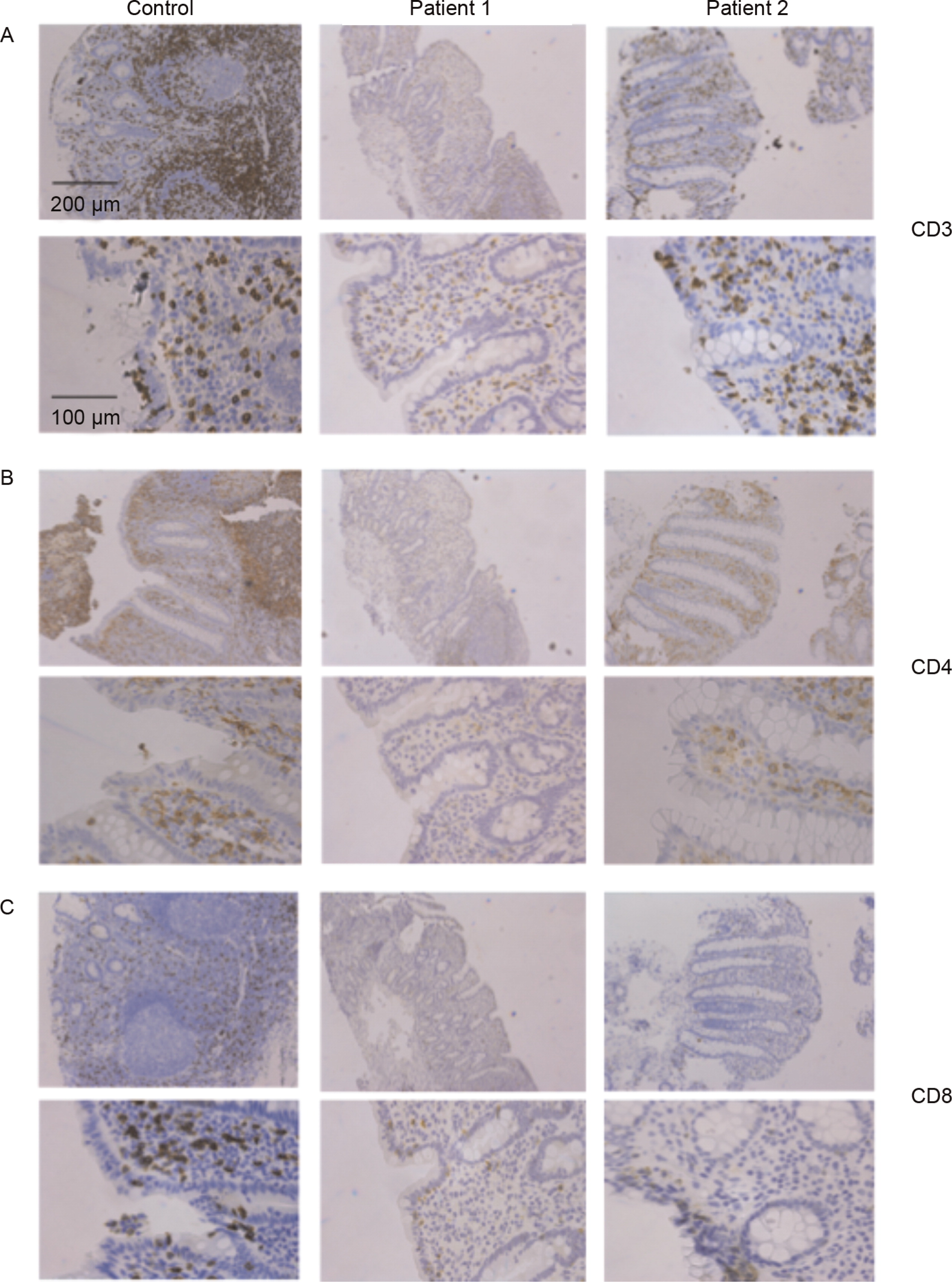
In patients with mutations in the DBD of STAT1, a progressive decrease in T-lymphocyte numbers has been demonstrated (Sharfe et al. 2014). We studied T-cell distribution in the gut mucosa of 2 of these patients. Colonic biopsy samples were stained for T-lymphocyte markers including CD3, CD4, and CD8. CD3 + T cells expressed in patient 2 were comparable with controls, whereas in patient 1 a marked decrease was detected within the intestinal mucosa, indicating that the underlying STAT1 mutation in patient 1 also affected gut T cells (Figure 2). Examining CD4 as well as CD8 showed diminished staining with a more prominent decrease in CD8 positive lymphocytes in patient 2 (Figure 2).
Figure 2:
Expression of total and activated STAT1 in enterocytes
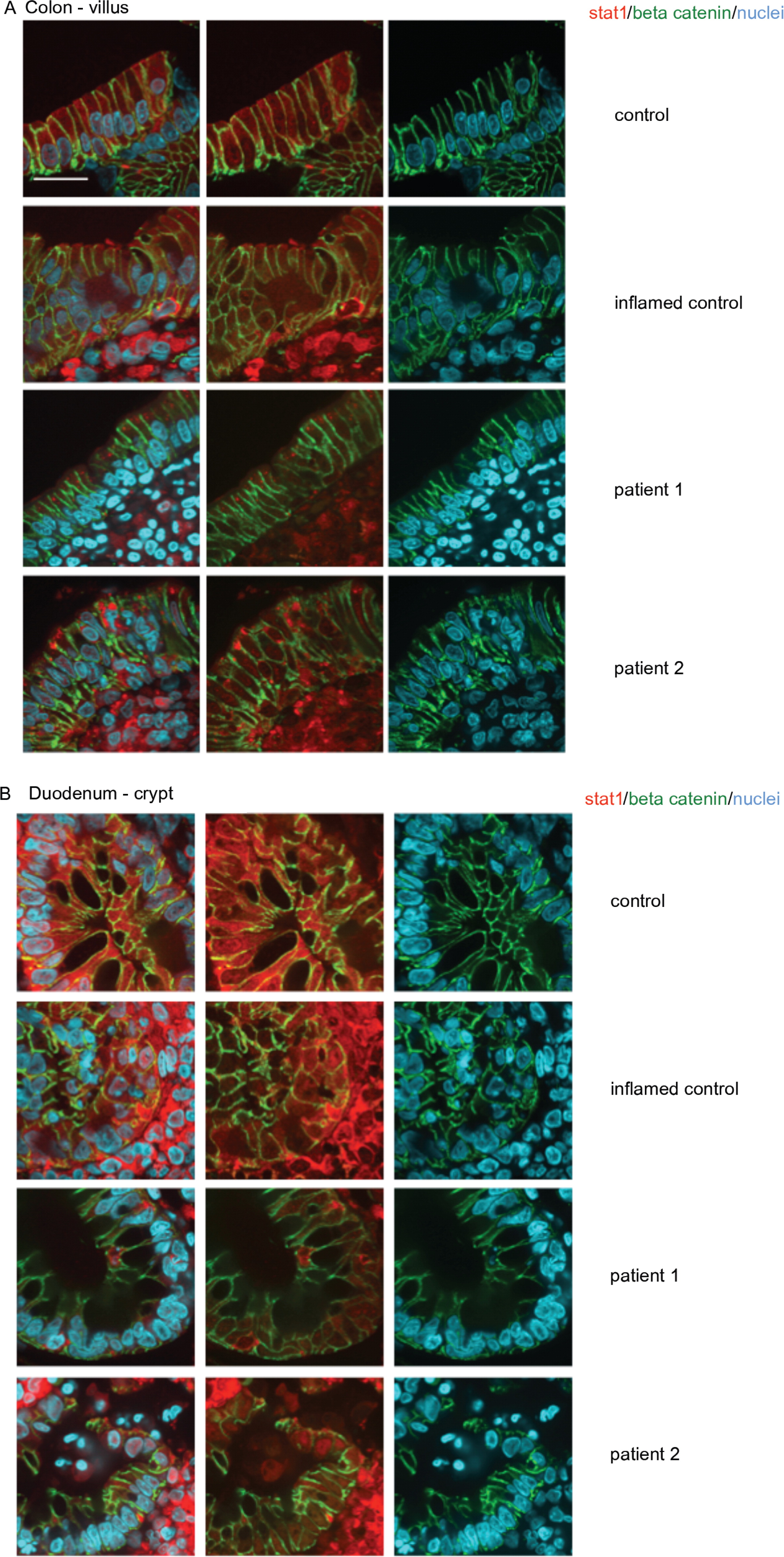
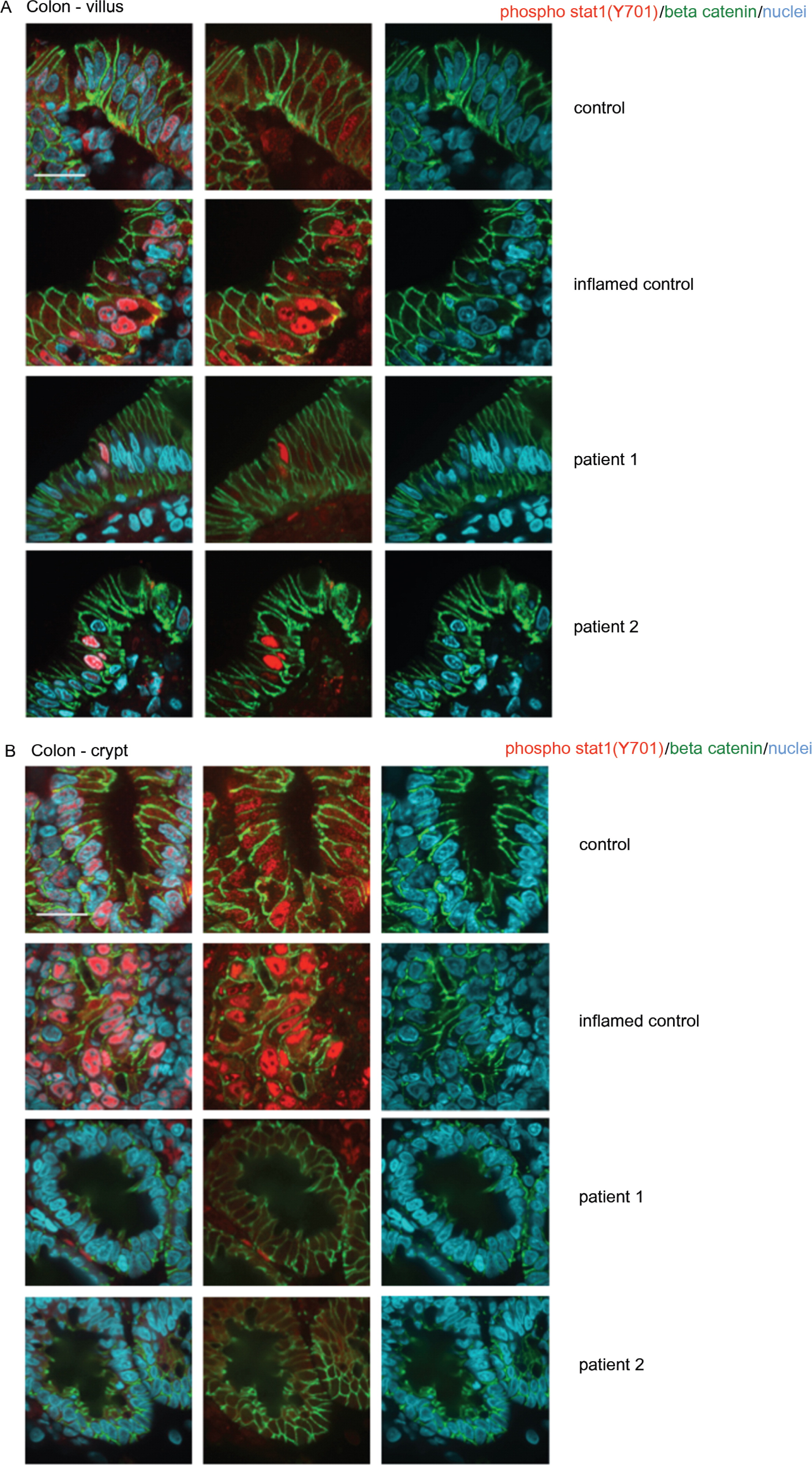
We next examined duodenal and colonic biopsy samples from the healthy controls, IBD controls, and both of the patients by labelling for total and activated STAT1. Additionally, cell–cell adhesion was analyzed using beta catenin staining. In the control samples, total STAT1 staining showed diffuse distribution throughout the cytoplasm of duodenal and colonic enterocytes (Figures 3 and 4). IBD controls demonstrated similar localization of STAT1, but the increased numbers of nonepithelial cells within the mucosa were predicted to be immune cells and activated by inflammatory responses as typically observed in inflammatory bowel disease (Figures 3 and 4). Analyses of STAT1 expression in duodenal and colonic enterocytes of both STAT1 deficiency patients showed that cytoplasmic STAT1 aggregates subapically; this was more predominantly observed in colonic biopsies from patient 2 (Figures 3 and 4). Those STAT1 aggregates were only observed in mature villous enterocytes, but not in immature crypt enterocytes (Figures 3 and 4). In duodenal and colonic crypt enterocytes of the STAT1 mutated patients, total STAT1 appeared to be reduced when compared with normal and IBD controls (Figures 3 and 4). Both STAT1 deficiency patients, as well as the IBD control, showed STAT1-positive cells staining within the mucosa of villous and crypts, they were predicted to be immune cells (Figures 3 and 4).
Figure 3:
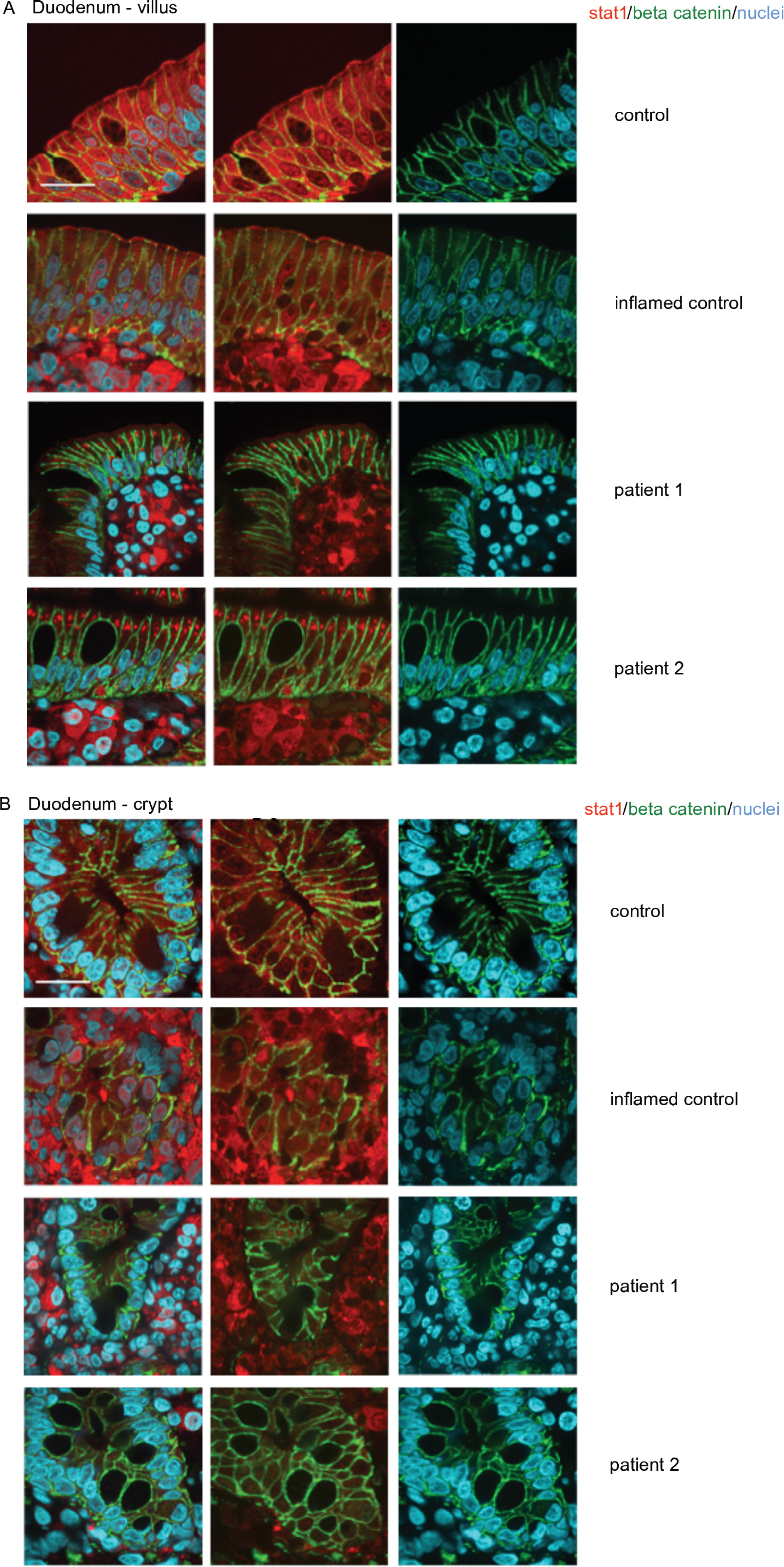
Figure 4:
We next determined if STAT1 activation by phosphorylation was affected in patient enterocytes by examining localization of phosphorylated STAT1 in both duodenal and colonic biopsy samples. Intriguingly, in IBD controls of duodenal and colonic biopsies, phospho-STAT1 was located in every enterocyte mostly within the nucleus suggesting that inflammatory responses activate STAT1-dependent pathways (Figures 5 and 6). In both patients, villous and crypts showed decreased numbers of enterocytes expressing phospho-STAT1 within the nucleus, indicating that the response to inflammation might be impaired due to the underlying STAT1 mutations (Figures 5 and 6). Consistently, both patients demonstrated changes in total and activated STAT1 expression within their intestinal epithelium.
Figure 5:
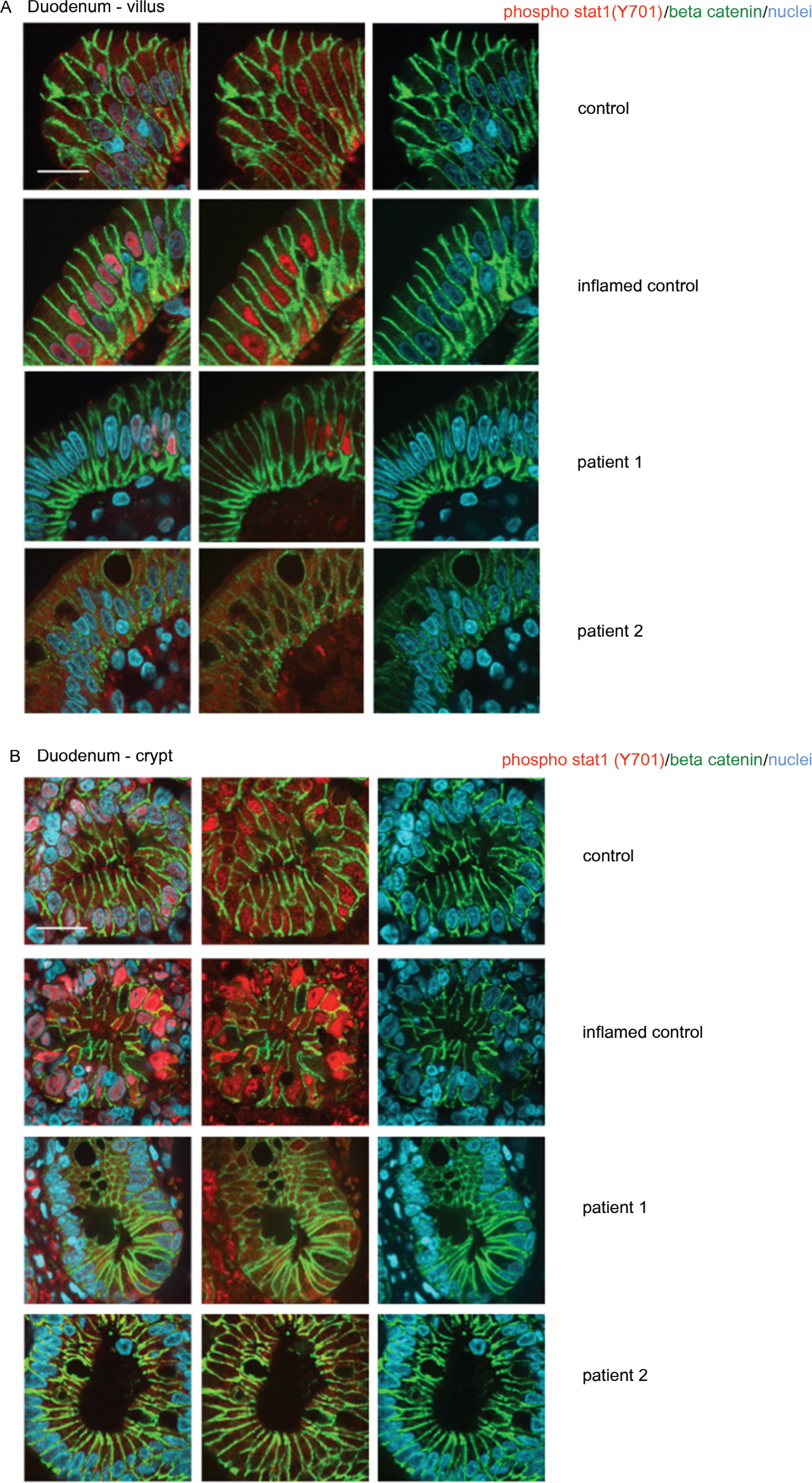
Figure 6:
Analysis of patient lymphoblasts
To further assess if STAT1 expression and activation is altered in patients, protein expression analysis was performed on patient-derived EBV-transformed lymphoblasts. As lymphoblasts of patient 1 were previously characterized (Sharfe et al. 2014), only data on lymphoblasts from patient 2 are presented here.
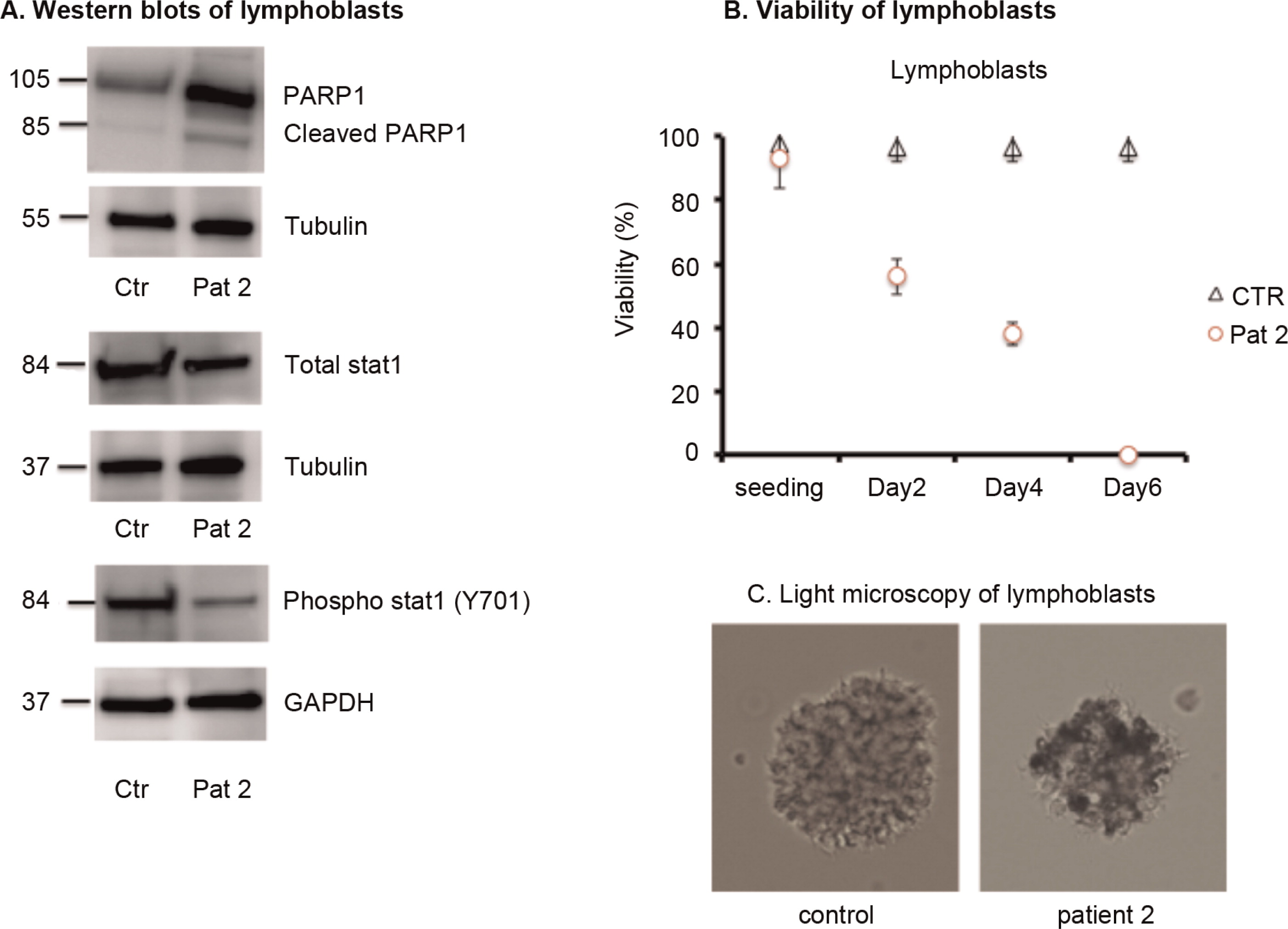
First we confirmed decreased total and activated STAT1 in patient 2, as observed in immunostainings of intestinal biopsies. Lymphoblasts of patient 2 showed decreased phospho-STAT1 expression when compared with controls, indicating impaired activation of STAT1 signalling (Figure 7). Next, differences in lymphoblast cell growth and morphology were detected when compared with controls. Cell viability was assessed after 2, 4, and 6 days from the initial cell seeding. On day 2 almost 40%, on day 4 it was 60%, and on day 6 virtually 100% of the lymphoblasts of patient 2 were dead indicating that the underlying STAT1 mutation drives lymphoblast apoptosis (Figure 7). Analysis of lymphoblast cell morphology with light microscopy showed that the cells of patient 2 had features of cell death when compared with controls. The control lymphoblasts showed clusters of bright, round cells, whereas in patient 2 the clusters consisted mostly of brownish cell debris, indicating that those lymphoblasts are undergoing apoptosis (Figure 7). To further confirm light microscopy features of cell death, the total and cleaved Poly (ADP-ribose)-Polymerase 1 (PARP1) expression levels were determined in lymphoblasts of patient 2 by immunoblot analysis. PARP1 is a nuclear DNA repair enzyme, activated by detection of single-strand DNA breaks in response to environmental stress and involved in apoptosis induction by serving as a target of many intra-cellular executer (ICE) caspases (Godon et al. 2008). Thereby, comparing PARP1 protein expression in the lymphoblasts of the controls with patient 2, demonstrated significantly increased expression of total PARP1, as well as the presence of cleaved PARP1, indicating that those lymphoblasts are undergoing programmed cell death (Figure 7). In summary, lymphoblasts of both patients with STAT1 dysfunction showed less viability and increased susceptibility to cell death.
Figure 7:
Discussion
Here we described 2 patients with mutations in the DBD of STAT1 presenting with recurrent bacterial and fungal infections as well as chronic diarrhea due to intestinal inflammation during infancy. It remains unclear why patients with heterozygous mutations in STAT1 develop severe colitis. It has been previously reported that increased activation of the STAT1 signalling pathway might play a role in the pathogenesis of inflammatory bowel disease (IBD), specifically in ulcerative colitis and chronic colonic inflammation (Podolsky et al. 1991; Schreiber et al. 2002). It has been proposed that the mucosal immune system, which normally acts in harmony to maintain a delicate balance between pro- and antiinflammatory cytokines, is disturbed in IBD (Podolsky et al. 1991; Wyatt et al. 1993; Schreiber et al. 2002). Indeed, predominance of pro-inflammatory cytokines has been observed in this condition. In addition, activated STAT1 signalling has been characterized in detail in ulcerative colitis (UC) (Schreiber et al. 2002). Further, altered activation of STAT1 in UC has been described as a consequence of lower levels of SOCs proteins, inhibitors of the JAK–STAT signalling cascade (Schreiber et al. 2002). Moreover, glucocorticoid treatment in those patients improved outcome and intestinal symptoms by altering STAT1 phosphorylation of immune cells in the intestinal mucosa via activation of SOCs proteins (Schreiber et al. 2002). In full agreement with these experiments, colitis in mouse models demonstrated similar patterns of increased activation in the STAT1 signalling by increased translocation of the phosphorylated STAT1 complex into the nucleus and increased activation of pro-inflammatory signalling pathways (Wu et al. 2012). Intriguingly, the 2 patients presented here had mutations in the DBD of the STAT1 gene, predicting impaired nuclear transcription of STAT1-related genes (Sharfe et al. 2014). It is not clear whether mechanisms underlying colitis in these patients are identical to IBD. Actually, both patients showed distinct features dissimilar to IBD. Histological changes of inflammation with increased mucosal infiltrates and architectural alterations within the villous and crypt epithelium were observed. Total STAT1 expression in enterocytes, STAT1 aggregated subapically in the cytoplasm of mature duodenal, and colonic villous enterocytes were markedly decreased in expression and activation in comparison with IBD. Further, phosphorylated STAT1, although present in the nucleus of colonic enterocytes of patients, was less prominent when compared with IBD control. Similarly EBV-transformed lymphoblasts derived from patients showed decreased levels as well as reduced phosphorylation of STAT1.
Other mechanisms may contribute to the development of colitis in these patients. Both patients showed decreased numbers of T lymphocytes within the intestinal mucosa. It has been previously shown that intestinal intraepithelial lymphocyte–enterocyte crosstalk was important to the production of antimicrobial proteins such as defensin, lysozyme, and angiogenin 4 by Paneth cells resulting in increased activity against grampositive and negative bacteria, protozoa, as well as fungi. This process prevents pathogen invasion within the intestinal mucosa and inflammation (Walker et al. 2013). Failure of this barrier contributes to development of colitis in IBD. The colitis in our patients could therefore be explained at least in part by dysregulation of gut immune cells. However, the striking unique pathological pattern in enterocytes (distinct from IBD), suggests that reduced STAT1 levels in these cells may lead to this phenotype as an independent factor. Indeed, primary changes in blood vessels, especially in the brain of patients with mutations in STAT1 have also been described (Grouhi et al. 1998; Nahum and Dalal 2014; Roifman 2014). Therefore, colitis in patients with heterozygous STAT1 mutation might be caused by a combination of decreased host defense and impaired STAT1 activation in gut enterocytes.
In conclusion, we reported here 2 patients carrying heterozygous STAT1 mutations who had features of chronic colitis. We found that the number of lymphocytes within the intestinal mucosa was decreased and uniquely STAT1 expression and activation in intestinal epithelial cells was altered. Both abnormalities may combine to determine host defense and lead to the inflammatory processes within the intestinal mucosa.
Acknowledgments
This work was supported by Immunodeficiency Canada, The Jeffrey Model Foundation, the Canadian Center for Primary Immunodeficiency and the Immunodeficiency Canada Distinguished Professor (CMR). This work was supported by the RESTRACOMP Award from the Research Institute of the Hospital for Sick Children, Toronto, Ontario. Thank you to Bernald Castro and Dr. Gino Somers, Division of Pathology, the Hospital for Sick Children, Toronto, Ontario, for providing us with technical assistance and patient material, respectively, as well as to the families for their participation in our studies. AMM is funded by a CIHR – Operating Grant (MOP119457) by the Leona M. and Harry B. Helmsley Charitable Trust to study VEOIBD.
REFERENCES
Boisson-Dupuis S., Kong X.F., Okada S., Cypowyj S., Puel A., Abel L., and Casanova J.L. Inborn errors of human STAT1: Allelic heterogeneity governs the diversity of immunological and infectious phenotypes Curr. Opin. Immunol 2012 24 4 364 -378
Chapgier A., Boisson-Dupuis S., Jouanguy E., Vogt G., Feinberg J., Prochnicka-Chalufour A., Casrouge A., Yang K., Soudais C., Fieschi C., Santos O.F., Bustamante J., Picard C., de Beaucoudrey L., Emile J.F., Arkwright P.D., Schreiber R.D., Rolinck-Werninghaus C., Rosen-Wolff A., Magdorf K., Roesler J., and Casanova J.L. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease PLoS Genet 2006 2 8 e131
Chapgier A., Kong X.F., Boisson-Dupuis S., Jouanguy E., Averbuch D., Feinberg J., Zhang S.Y., Bustamante J., Voft G., Lejeune J., Mayola E., de Beaucoudrey L., Adel L., Engelhard D., and Casanova J.L. A partial form of recessive STAT1 deficiency in humans J. Clin. Invest 2009 119 1502 -1514
Dupuis S., Dargemont C., Fieschi C., Thomassin N., Rosenzweig S., Harris J., Holland S.M., Schreiber R.D., and Casanova J.L. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation Science 2001 293 5528 300 -303
Dupuis S., Jouanguy E., Al-Hajjar S., Fieschi C., Al-Mohsen I.Z., Al-Jumaah S., Yang K., Chapgier A., Eldenschenk C., Eid P., Al Ghonaium A., Tufenkejh H., Frayha H., Al-Gazlan S., Al-Rayes H., Schreiber R.D., Gresser I., and Casanova J.L. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency Nat. Genet 2003 33 3 388 -391
Faitelson Y., Bates A., Shroff M., Grunebaum E., Roifman C.M., and Naqvi A. A mutation in the STAT1 DNA-binding domain associated with hemophagocytic lymphohistocytosis LymphoSign J 2014 1 2 87 -95
Godon C., Cordelières F.P., Biard D., Giocanti N., Mégnin-Chanet F., Hall J., and Favaudon V. PARP inhibition versus PARP-1 silencing: Different outcomes in terms of single-strand break repair and radiation susceptibility Nucleic Acids Res 2008 36 13 4454 -4464
Grouhi M., Dalal I., Nisbet-Brown E., and Roifman C.M. Cerebral vasculitis associated with chronic mucocutaneous candidiasis J. Pediatr 1998 133 4 571 -574
Liu L., Okada S., Kong X.F., Kreins S.C., Abhyankar A., Toubiana J., Itan Y., Audry M., Nitschke P., Masson C., Toth B., Flatot J., Migaud M., Chrabieh M., Kochetkov T., Bolze A., Borghesi A., Toulon A., Hiller J., Eyerich S., Eyerich K., Gulácsy V., Chernyshova L., Chernyshov V., Bondarenko A., Grimaldo R.M., Blancas-Galicia L., Beas I.M., Roesler J., Magdorf K., Engelhard D., Thumerelle C., Burgel P.R., Hoernes M., Drexel B., Seger R., Kusuma T., Jansson A.F., Sawalle-Belohradsky J., Belohradsky B., Jouanguy E., Bustamante J., Bué M., Karin N., Wildbaum G., Bodemer C., Lortholary O., Fischer A., Blanche S., Al-Muhsen S., Reichenbach J., Kobayashi M., Rosales F.E., Lozano C.T., Kilic S.S., Oleastro M., Etzioni A., Traidl-Hoffmann C., Renner E.D., Abel L., Picard C., Maródi L., Boisson-Dupuis S., Puel A., and Casanova J.L. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis J. Exp. Med 2011 208 1635 -1648
Maródi L., Cypopwyj S., Tóth B., Smeekens S.P., Joosten L.A.B., Gilissen C., Arts P., Rosentul D.C., Carmichael A.J., Smits-van der Graaf C.A.A., Bart Jan Kullberg B.J., van der Meer J.W.M., Desa Lilic D., Veltman J.A., and Netea M.G. Molecular mechanisms of mucocutaneous immunity against candida and Staphylococcus species J. Allergy Clin. Immunol 2012 130 1019
Nahum A. and Dalal I. Clinical manifestations associated with novel mutations in the coiled-cloi domain of STAT1 LymphoSign J 2014 1 2 97 -103
Najjar I. and Fagard R. STAT1 and pathogens, not a friendly relationship Biochimie 2010 92 5 425 -444
Ngan B., Merico D., Maruc N., Kim V.H.D., Upton J., Bates A., Herbrick J., Nalpathamkalam T., Thiruvahindrapuram B., Cox P., and Roifman C.M. Mutations in tetratricopeptide repeat domain 7A (TTC7A) are associated with combined immunodeficiency with dendriform lung ossification but no intestinal atresia LymphoSign J 2014 1 10 -β26
O'Shea J.J., Holland S.M., and Staudt L.M. JAKs and STATs in immunity, immunodeficiency, and cancer N. Engl. J. Med 2013 368 2 161 -170
Podolsky D.K. Inflammatory bowel disease (1) N. Engl. J. Med 2002 347 6 417 -429
Roifman C.M. Monallelic STAT1 mutations and disease patterns LymphoSign J 2014 1 2 57 -59
Roifman C.M., Somech R., Kavadas F., Pires L., Nahum A., Dalal I., and Grunebaum E. Defining combined immunodeficiency J. Allergy Clin. Immunol 2012 130 1 177 -183
Schreiber S., Rosenstiel P., Hampe J., Nikolaus S., Groessner B., Schottelius A., Kühbacher T., Hämling J., Fölsch U.R., and Seegert D. Activation of signal transducer and activator of transcription (STAT) 1 in human chronic inflammatory bowel disease Gut 2002 51 3 379 -385
Sharfe N., Nahum A., Newell A., Dadi H., Ngan B., Pereira S.L., Herbrick J.A., and Roifman C.M. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1 J. Allergy Clin. Immunol 2014 133 3 807 -817
Takezaki S., Yamada M., Kato M., Park M.J., Maruyama K., Yamazaki Y., Chida N., Ohara O., Kobayashi I., and Ariga T. Chronic Mucocutaneous candidiasis caused by a gan-of-function mutation in the STAT1 DNA-binding domain J. Immunol 2012 189 1521 -1526
Uzel G., Sampaio E.P., Lawrence M.G., Hsu A.P., Hackett M., Dorsey M.J., Noel R.J., Verbsky J.W., Freeman A.F., Janssen E., Bonilla F.A., Pechacek J., Chandrasekaran P., Browne S.K., Agharahimi A., Gharib A.M., Mannurita S.C., Yim J.J., Gambineri E., Torgerson T., Tran D.Q., Milner J.D., and Holland S.M. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome J. Allergy Clin. Immunol 2013 131 6 1611 -1623
van de Veerdonk F.L., Plantinga T.S., Hoischen A., Smeekens S.P., Joosten L.A., Gilissen C., Arts P., Rosentul D.C., Carmichael A.J., Smits-van der Graaf C.A., Kullberg B.J., van der Meer J.W., Lilic D., Veltman J.A., and Netea M.G. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis N. Engl. J. Med 2011 365 1 54 -61
Walker C.R., Hautefort I., Dalton J.E., Overweg K., Egan C.E., Bongaerts R.J., Newton D.J., Cruickshank S.M., Andrew E.M., and Carding S.R. Intestinal intraepithelial lymphocyte-enterocyte crosstalk regulates production of bactericidal angiogenin 4 by Paneth cells upon microbial challenge PLoS One 2013 8 12 e84553
Wu X., Guo W., Wu L., Gu Y., Gu L., Xu S., Wu X., Shen Y., Ke Y., Tan R., Sun Y., and Xu Q. Selective sequestration of STAT1 in the cytoplasm via phosphorylated SHP-2 ameliorates murine experimental colitis J. Immunol 2012 189 7 3497 -3507
Wyatt J., Vogelsang H., Hübl W., Waldhöer T., and Lochs H. Intestinal permeability and the prediction of relapse in Crohn's disease Lancet 1993 341 8858 1437 -1439
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 1 • March 2016
Pages: 19 - 33
History
Received: 22 September 2015
Accepted: 14 November 2015
Version of record online: 19 December 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Cornelia Thoeni, Ellen A. Hamilton, Abdul Elkadri, Ryan Murchie, Karoline Fiedler, Adi Ovadia, Nigel Sharfe, Bo Ngan, Ernest Cutz, Amit Nahum, Aleixo M. Muise, and Chaim M. Roifman. 2016. The effects of STAT1 dysfunction on the gut. LymphoSign Journal.
3(1): 19-33. https://doi.org/10.14785/lpsn-2015-0012
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
