Cartilage–hair hypoplasia: a spectrum of clinical and radiological findings
Abstract
Introduction: Cartilage–hair hypoplasia (CHH) is a rare skeletal dysplasia that presents with various degrees of immunodeficiency, short stature, and a susceptibility to malignancies. Individuals with CHH can present with severe combined immunodeficiency or combined immunodeficiency and are at risk for severe and unusual infections irrespective of their laboratory findings. In addition, individuals with CHH can present with variable skeletal abnormalities, mainly involving the metaphysis of long bones. CHH is a rare disease and familiarity with the variable features is crucial for diagnosis.
Methods: We report the clinical, radiological, and genetic findings for 5 patients with proven diagnoses of CHH.
Results: In this study we describe a cohort of patients with CHH and present their clinical findings and progressions. In addition, we present the radiological images and the immunological investigations that were done in these patients. Although all the patients in our cohort had poor cellular immunity, they had a variable clinical course. Three out of 5 patients received a bone marrow transplant (BMT) and 2 out of 5 died at an early age (1 after BMT). Those who had poor humoral function had a worse prognosis compared with those with good humoral function. The skeletal findings were characteristic for CHH.
Conclusion: CHH is a disease with a variable presentation. Clinicians should be aware of the characteristic skeletal and immunological findings to identify the disease as early as possible.
Statement of novelty: We present novel clinical and radiological findings in patients with variable RMRP gene mutations.
Introduction
Cartilage–hair hypoplasia (CHH) is a rare skeletal dysplasia inherited as an autosomal recessive trait. It was first described among the Old Order Amish and the Finnish population (McKusick et al. 1965) and it is caused by mutations in the ribonuclease mitochondrial RNA-processing (RMRP) gene (Sulisalo et al. 1993; Ridanpää et al. 2001). The syndrome's hallmarks include various degrees of immunodeficiency, short stature, fine sparse hair, Hirschsprung disease, and a susceptibility to hematologic malignancies. The major radiographic findings in CHH are short metacarpals and phalanges; short, wide iliac wings; short, thick long bones; femoral bowing; and metaphyseal dysplasia (Kwan et al. 2012).
CHH is caused by mutations in the RMRP (RNA component of mitochondrial RNA processing endoribonuclease) gene that encodes for the untranslated RNA component of a mitochondrial RNA-processing endonuclease, a protein-RNA complex that plays a role processing of ribosomal RNA (rRNA), cleavage of mitochondrial RNA (mRNA), and in cell cycle control. The most prevalent mutation among the Finnish and the Amish is the 70 A>G (Thiel et al. 2007). In North America most cases are caused by compound herterozygous mutations that commonly affect the gene's regulatory elements (Notarangelo et al. 2008).
CHH can be diagnosed clinically in an individual with short stature, disproportionally short limbs, and fine hair, but confirmation of the diagnosis should be done by genetic analysis of the RMRP gene. Individuals with CHH can present with severe combined immunodeficiency (SCID) or combined immunodeficiency (CID) and are at risk for severe and unusual infections irrespective of their laboratory findings. Other complications also include malignancy, and autoimmunity. An early diagnosis of CHH is crucial to prevent morbidity and mortality from these immune-related disorders.
The short stature and radiological features are frequently the first clue for the diagnosis. However, recognition of the typical skeletal findings may be delayed as most clinicians are rarely familiar with CHH.
Therefore, we present here the clinical and radiological features of 5 patients with RMRP mutations.
Case 1
Patient 1, a female with an Ashkenazi Jewish background, was born at term to healthy parents with no known consanguinity. Although her birth weight and head circumference were around the 50th percentile, she was relatively short, as her length was below the 10th percentile. Soon after birth she was described as having fine and sparse hair, short proximal limbs, prominent occiput, overlapping of the digits, and a small umbilical hernia. The tibial and humeri length were equivalent to 30 and 29 weeks gestational age, respectively. At the age of 6 months she was found to have low IgG and was referred to the immunology clinic. At that age she didn't suffer from severe or recurrent infections and she showed only mild developmental delay of her gross motor skills. A skeletal survey showed metaphyseal cupping/scalloping in the distal femora, radial and ulna, as well as metacarpal foreshortening (Figures 1 and 2).
Figure 1:
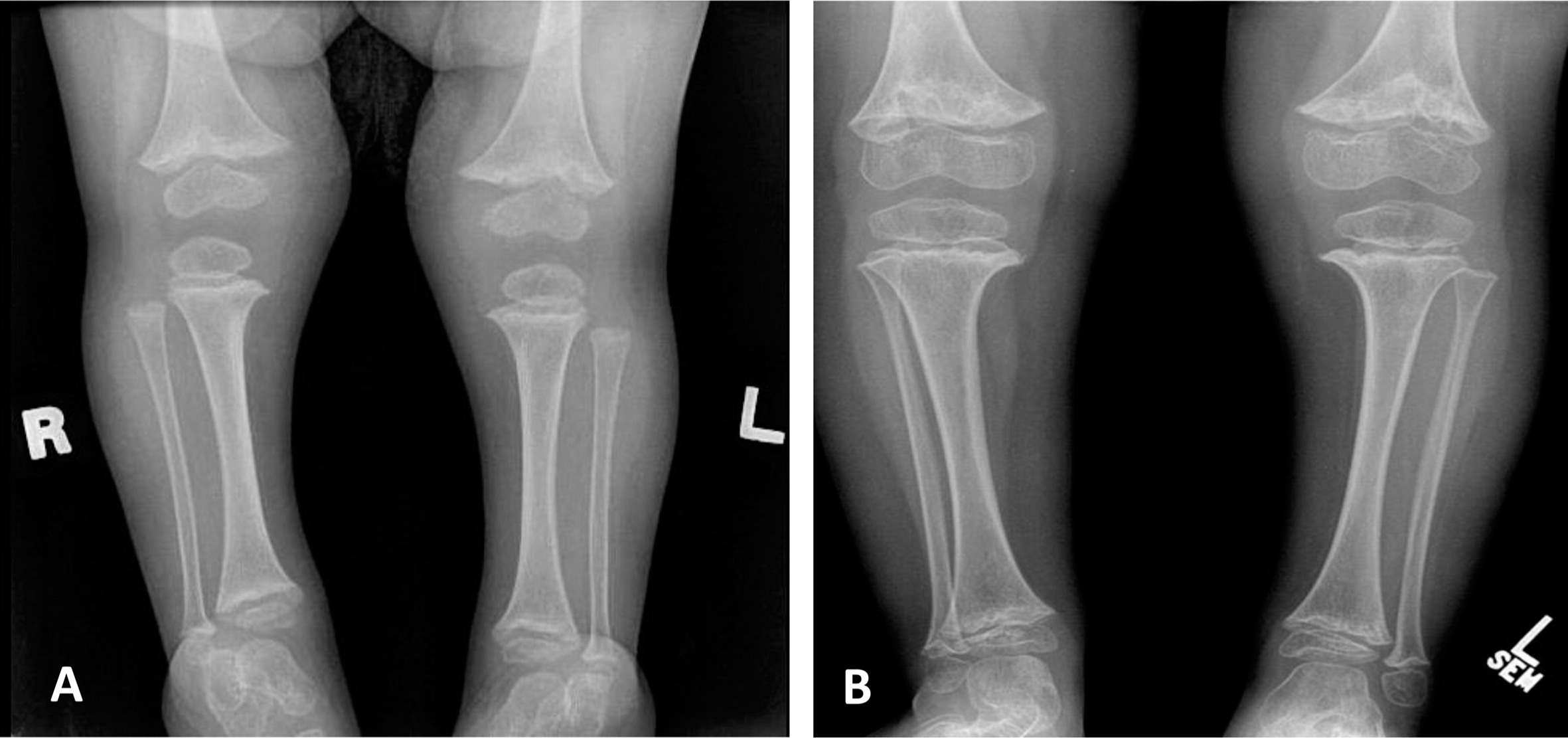
Figure 2:
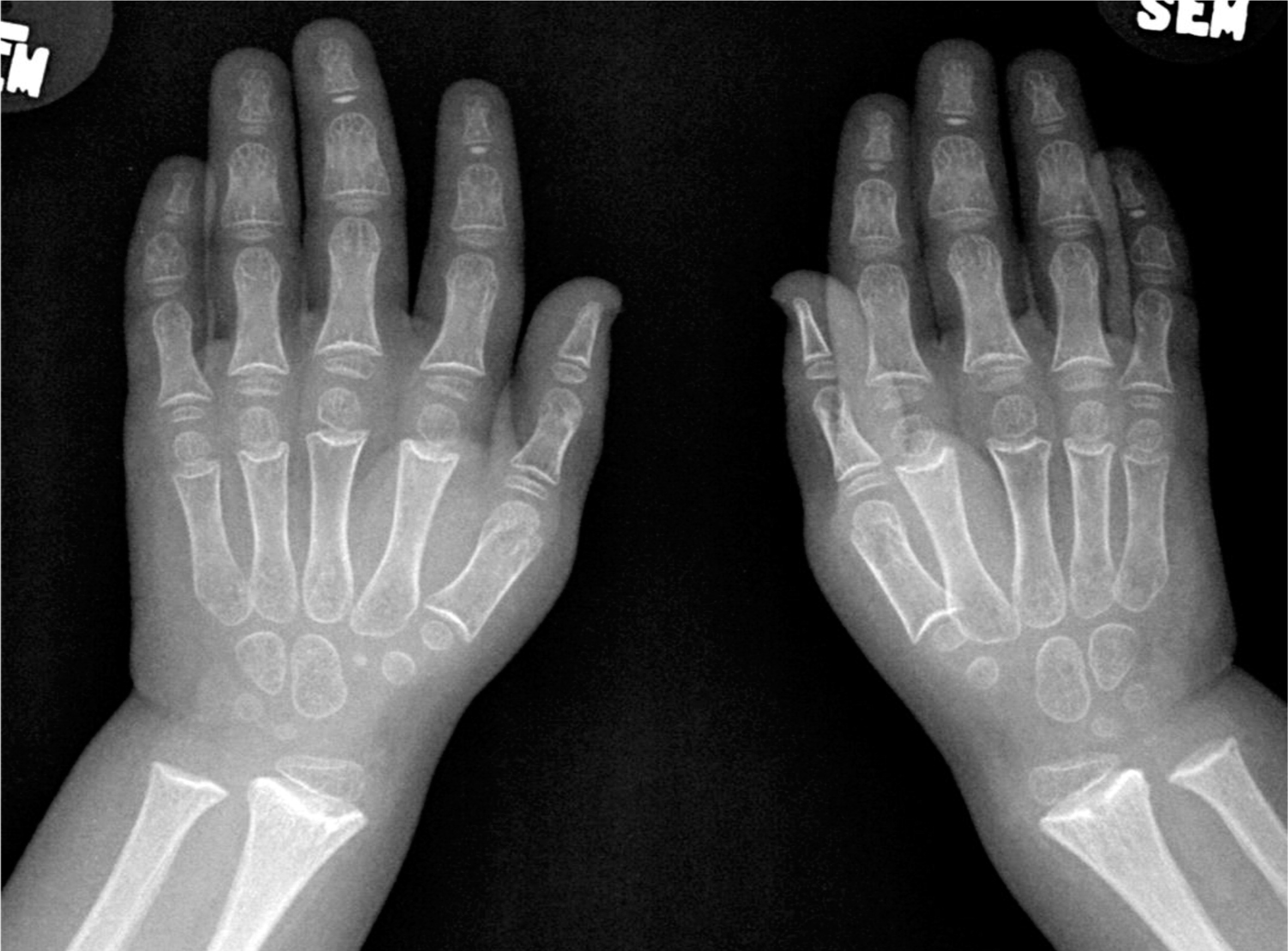
The initial immunological workup showed a SCID phenotype of T−B+NK+, absence of the T-cell receptor excision circle (TREC), poor T-cell function, and a low T-cell number.
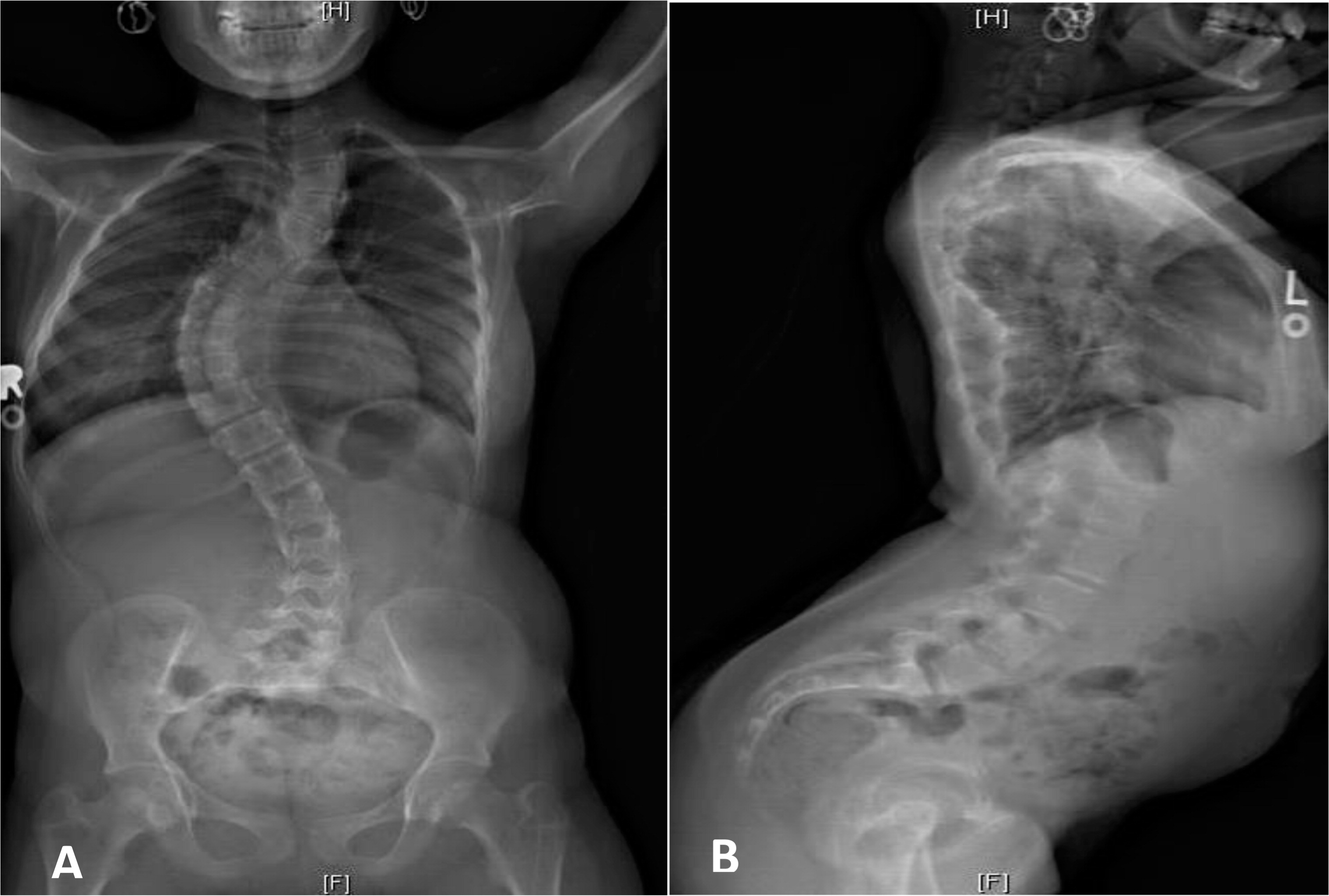
At the age of 8 months the patient was diagnosed with CHH based on a mutation analysis that showed the common mutation 70 A>G in the RMRP gene. She received a bone marrow transplant (BMT) from a matched unrelated donor at the age of 14 months after full myeloablative conditioning (Busulfan and Cyclophosphamide) and was engrafted shortly after with no complications. Other than severe scoliosis that developed in the 2nd decade of her life (Figure 3), her long-term engraftment and immune reconstitution remain excellent.
Figure 3:
Case 2
Patient 2, a female of French–Irish and Native American origin, was born at term with no complications. Her older sibling was diagnosed with CHH and her antenatal fetal ultrasound showed short limbs at 27 weeks of gestation. Genetic work-up demonstrated a mutation in the RMRP gene 70 A>G and she was diagnosed with CHH at the age of 1 month. At the age of 10 days she suffered from respiratory syncytial virus pneumonia but recovered after 2 weeks. The immune work-up in the first few months of life showed low a TREC number, T- and B-cell lymphopenia, normal levels of immunoglobulins, and poor T-cell function. She also had intermittent neutropenia that eventually resolved spontaneously. Further evaluation showed good antibody responses to vaccination with tetanus and pneumovax, although her T-cell function continued to be poor. Clinically, she suffered from constipation in the first 3 years of life but she didn't have severe or recurrent infection or signs of autoimmunity. Currently she is doing well and treated only with dapsone as a Pneumocystis jiroveci pneumonia prophylaxis.
Case 3
Patient 3, a female with an English background, was born at term to healthy parents with no known consanguinity. Family history was remarkable only for a maternal nephew with myotubular myopathy. Her birth weight was 3.9 kg but she failed to thrive soon after birth. With the exception of her failure to thrive, she was doing well until the age of 2 years when she started to suffer from recurrent episodes of pneumonia, and eventually she developed bronchiectasis by the age of 6 years. She was then referred to immunology for further evaluation. Immunological workup showed a low T-cell number, poor T-cell function, and poor response to pneumococcal vaccine. She started treatment with IVIG and prophylaxis antibiotic. Skeletal films showed the characteristic skeletal changes in the metacarpals, distal ulna (Figure 4), and distal fibula.
Figure 4:
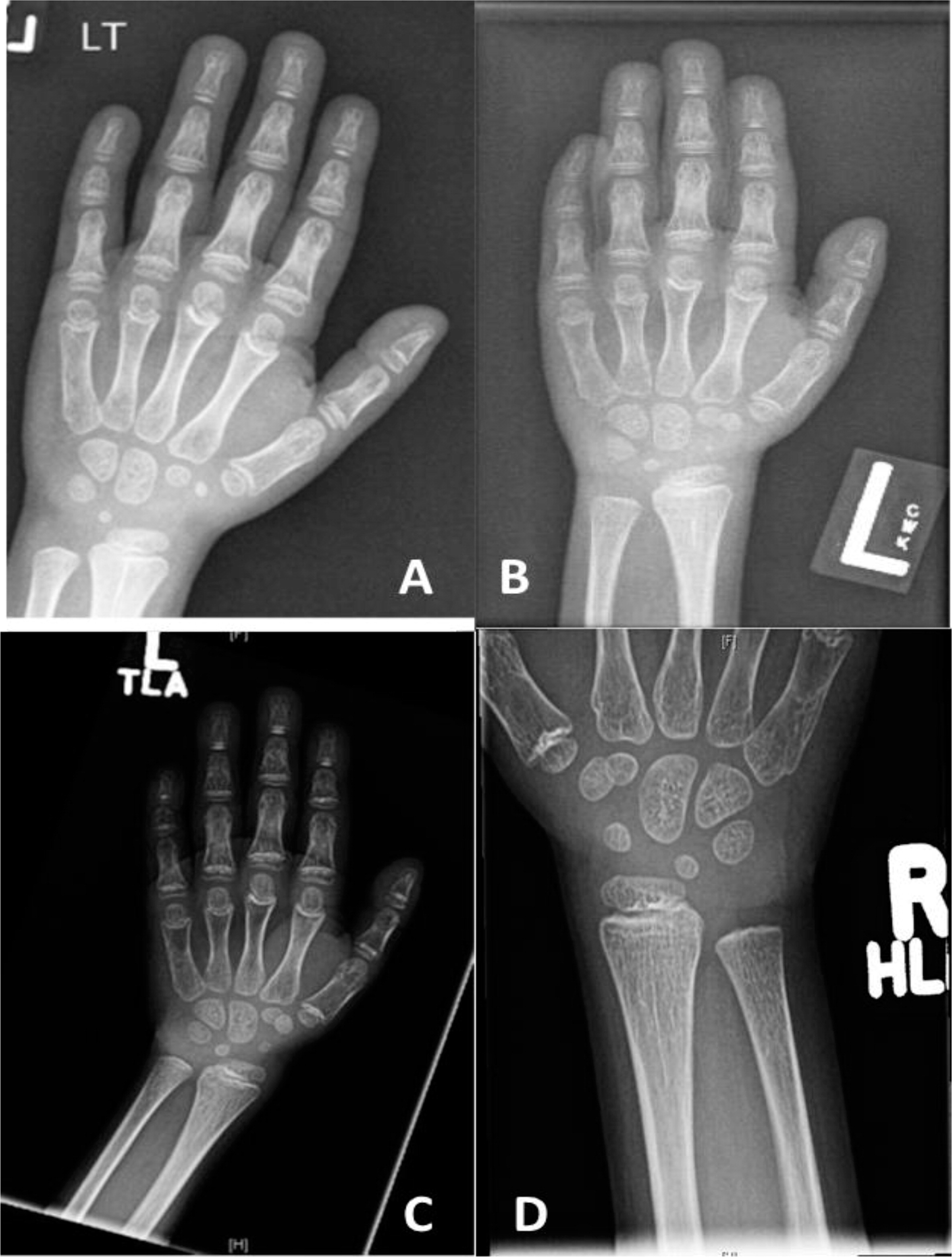
Genetic analysis confirmed the diagnosis of CHH by identifying 2 mutations in the RMRP gene 146 G>A/242 A>G, each donated by the father and mother, respectively. During childhood she suffered from autoimmune hemolytic anemia. BMT was not done owing to the lack of an appropriate donor. At the age of 12 years she presented with acute enchephalitis and cognitive decline. She was treated with a broad spectrum antibiotic and anti-viral medication and eventually with immunosuppression after an infectious etiology was excluded. Unfortunately, she didn't recover and passed away a few months later.
Case 4
Patient 4, a male from French–Irish and Native American origin, was born at term with a weight of 3.4 kg. At 30 weeks of gestation, an ultrasound revealed small long bones and a bell-shaped chest. An amniocentesis was not diagnostic but genetic investigation after birth confirmed the diagnosis of CHH at the age of 7 months with the finding of the common mutation 70 A>G in the RMRP gene. Other features of CHH included fine sparse hair, constipation (with no evidence of Hirschsprung disease), and bowed legs (Figure 5).
Figure 5:
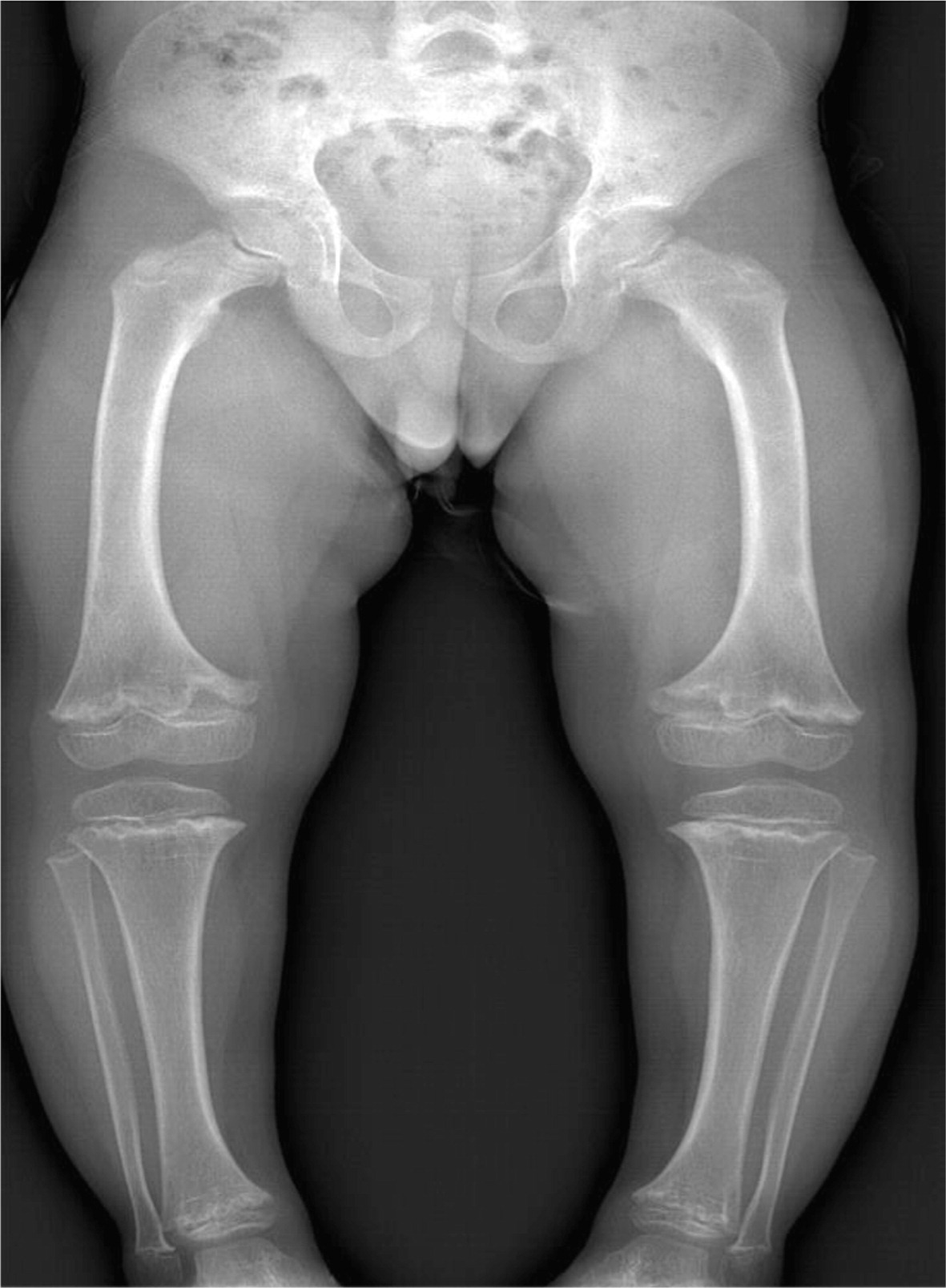
His immunological investigation showed a normal level of immunoglobulins as well as adequate levels of antibodies to vaccination, but his T-cell number and function were poor on several occasions. At the age of 4 years, his bloodwork showed a restricted T-cell repertoire with clonal expansion of the VB12 and VB13 families for CD4+ cells and expansion of the VB1 family for CD8+ cells. Interestingly, the patient didn't have any severe or recurrent infections, nor did he suffer autoimmune manifestations.
Considering the patient's poor T-cell function, he had a BMT from a matched sibling donor at the age of 4.5 years. Following the transplant, at first he developed only minor acute skin graft versus host disease (GVHD), which was controlled with immunosuppression. However, a year later, he was diagnosed with chronic skin GVHD. The rash subsided with a course of steroids and methotrexate, but since then he continues to have contractures of the hands and feet. The patient continues to function well, engraftment is complete, and long-term immune reconstitution is solid.
Case 5
Patient 5, a male, was born to consanguineous parents of French Canadian origin. His birth height was below the 3rd percentile while his weight was at the 25th percentile. At the age of 8 weeks he developed Omenn syndrome with generalized erythroderma, lymphadenopathy, diarrhea, and irritability. His immune work-up showed low levels of immunoglobulins, clonal expansion of the CD4+ T cell, and absent CD8+ T cells. In addition, in vitro responses to mitogens were poor.
Genetic analysis revealed 2 mutations in the RMRP gene, 154 G>C and 15 nucleotide duplication at position −25, each donated by the mother and father, respectively. At the age of 8 months he received a matched unrelated donor BMT but developed severe GVHD post-transplant. He suffered from severe skin involvement of his GVHD, acute respiratory distress syndrome with pulmonary vasculopathy, hypertension, renal failure, and secondary hypertrophic cardiomyopathy. He was treated with multiple agents against GVHD but continued to have recurrent exacerbations of GVHD and deterioration of his cardiorespiratory status. He died at the age of 3 years and 4 months from multi-organ failure.
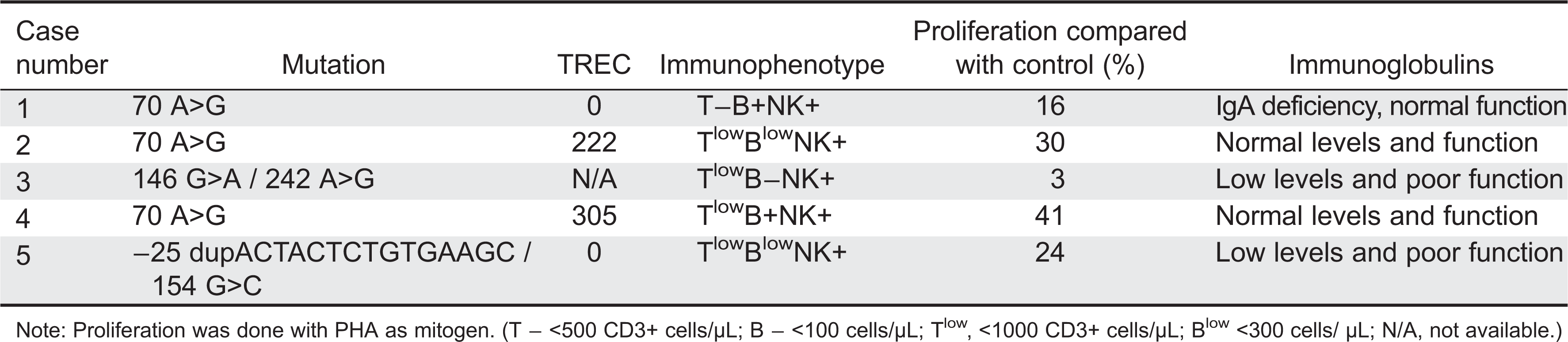
Table 1 summarizes the immunological findings of Cases 1–5.
Table 1:
Discussion
CHH is a rare autosomal recessive skeletal dysplasia with severe short stature; characteristic skeletal changes affecting mainly the metaphysis; and extraskeletal features involving the immune, hematologic, and the gastrointestinal system. The phenotypic features correlate with the degree of reduced rRNA or mRNA cleavage. RMRP mutations that reduce rRNA cleavage are associated with bone dysplasia, whereas defects that decrease mRNA cleavage are associated with hair hypoplasia, immunodeficiency, and hematologic abnormalities (Thiel et al. 2007; Notarangelo et al. 2008). The characteristic skeletal changes seen in CHH involve mainly the metaphysis with the findings of metaphyseal splaying, coarse irregularity, and lucent “cyst”-like lesions most prominent in the knees and ankles (Conwell et al. 2008). The epiphysis can also show some changes with rounding and a globular appearance. In addition, many other skeletal findings have been described including short metacarpals and phalanges; lumbar lordosis; short, wide iliac wings; femoral bowing; and scoliosis. Mild scoliosis was observed in one-fourth of the patients in 1 study and the incidence increased with age (Mäkitie et al. 1992). Indeed, 1 of our patients developed scoliosis in the 2nd decade of life in accordance with previous studies (McKusick et al. 1965; Ray et al. 1975).
The skeletal findings in CHH are not only variable but can also be misleading as they are not always evident in infancy (Glass and Tifft 1999) and might even disappear after a short period of time (Kwan et al. 2012). The characteristic metaphyseal irregularities and cystic lesions typically do not appear until after 2 years of age. After the epiphysis closes, the metaphysis may remain flared, angulated, and eroded (Mäkitie and Kaitila 1993) or may be normal (van derBurgt et al. 1991; Makitie et al. 1995). Our patients demonstrate the variable skeletal findings with classical metaphyseal changes and metacarpal foreshortening.
Although metaphyseal changes are the most common skeletal finding in patients with CHH, they are not restricted to this disorder as some other primary immunodeficiencies (PID) may present with multiple skeletal finding. Patients with Adenosine Deaminase Deficiency can develop different skeletal abnormalities with metaphyseal changes including prominent costochondral junctions (rachitic rosary), flaring of the anterior rib ends, pelvic dysplasia, squared off inferior scapula, or flattened vertebral bodies (platyspondyly) (Cederbaum et al. 1976). Patients with Schwachman–Diamond syndrome can present with metaphyseal chondrodysplasia in addition to bone marrow failure and pancreatic insufficiency (Mäkitie et al. 2004). Patients with Roifman immunoskeletal syndrome (spondyloenchondrodysplasia with immune dysregulation) have been described with metaphyseal and vertebral bone lesions with immune dysfunction and autoimmunity (Roifman and Melamed 2003). In addition, there are some reports of metaphyseal erosions in patients with deficiency of IL-1 receptor antagonist, a rare PID characterized with bone and skin abnormalities (Aksentijevich et al. 2009). It is important to mention that metaphyseal changes are not the only skeletal abnormalities seen in PID. Other skeletal changes such as spondyloepiphyseal dysplasia can be seen in Schimkes immuno-osseous dysplasia (short-trunk dwarfism as a result of spondyloepiphyseal dysplasia and immunologic abnormalities) (Hunter et al. 2010) and in Roifman syndrome (spondyloeiphyseal dysplasia, microcephaly, intrauterine growth restriction, growth retardation, and humoral immunodeficiency) (Roifman 1999).
Similar to the variability in the skeletal finding, the immune system in CHH can display a mild array of findings (Kavadas et al. 2008). Abnormal cellular immunity is present in the majority of CHH patients (van der Burgt et al. 1991; Mäkitie and Kaitila 1993), although there may be imperfect correlation between laboratory findings and susceptibility to infections (Makitie et al. 1998). In the Amish population, only 32% of the patients had severe or recurrent infections and only 8% of these children underwent BMT for CID (Rider et al. 2009). Irrespective of clinical phenotype, most patients in that study had lymphopenia and reduced lymphocyte proliferation in response to mitogens. In our small cohort, all the patients showed poor T-cell function with poor response to mitogen and a low TREC number, but only 2 patients had poor humeral function. Three out of the 5 patients had a BMT and 2 out of the 5 passed away in early age (one from GVHD post-BMT and one from encephalitis). Interestingly, the 2 patients who passed away in our cohort and the patients who needed BMT in the study by Rider et al. (2009) are the patients with the poor humeral function, a finding that suggests that poor humeral function is a bad prognostic factor in CHH. One of our patients presented with Omenn syndrome (Roifman et al. 2006), which is an unusual presentation of CHH that proves mutations in the RMRP RNA gene might be associated with a more complex presentation than the classical one. Involvement of other systems such as the gastrointestinal system has been described in CHH patients (mainly Hirschprung's disease), but was not common in our cohort. Two of our patients had a history of constipation but none needed significant intervention. Malignancy was not seen in our cohort, but our patients are still young and this complication may appear later in life.
In summary, we have illustrated the skeletal changes typical of CHH. When present they should trigger a thorough assessment of the immune system, especially when such patients suffer repeated infections.
REFERENCES
Aksentijevich I., Masters S.L., Ferguson P.J., Dancey P., Frenkel J., van Royen-Kerkhoff A., Laxer R., Tedgård U., Cowen E.W., Pham T.-H., Booty M., Estes J.D., Sandler N.G., Plass N., Stone D.L., Turner M.L., Hill S., Butman J.A., Schneider R., Babyn P., El-Shanti H.I., Pope E., Barron K., Bing X., Laurence A., Lee C.R., Chapelle D., Clarke G.I., Ohson K., Nicholson M., Gadina M., Yang B., Korman B.D., Gregersen P.K., Van Hagen P.M., Hak A.E., Huizing M., Rahman P., Douek D.C., Remmers E.F., Kastner D.L., and Goldbach-Mansky R. An autoinflammatory disease with deficiency of the interleukin-1–receptor antagonist N. Engl. J. Med. 2009 360 23 2426 -2437
Cederbaum S.D., Kaitila I., Rimoin D.L., and Stiehm E.R. The chondro-osseous dysplasia of adenosine deaminase deficiency with severe combined immunodeficiency J. Pediatr. 1976 89 5 737 -742
Conwell L.S., Hermanns P., and Zanki A. Short stature and metaphyeal dysplasia due to cartilage-hair hypoplasia J. Pediatr. Endocrinol. Metab. 2008 21 209 -211
Glass R.B.J. and Tifft C.J. Radiologic changes in infancy in McKusick cartilage hair hypoplasia Am. J. Med. Genet. 1999 86 4 312 -315
Hunter K.B., Lücke T., Spranger J., Smithson S.F., Alpay H., André J.-L., Asakura Y., Bogdanovic R., Bonneau D., Cairns R., Cransberg K., Fründ S., Fryssira H., Goodman D., Helmke K., Hinkelmann B., Lama G., Lamfers P., Loirat C., Majore S., Mayfield C., Pontz B.F., Rusu C., Saraiva J.M., Schmidt B., Shoemaker L., Sigaudy S., Stajic N., Taha D., and Boerkoel C.F. Schimke immunoosseous dysplasia: Defining skeletal features Eur. J. Pediatr. 2010 169 7 801 -811
Kavadas F.D., Giliani S., Gu Y., Mazzolari E., Bates A., Pegoiani E., Roifman C.M., and Notarangelo L.D. Variability of clinical and laboratory features among patients with ribonuclease mitochondrial RNA processing endoribonuclease gene mutations J. Allergy Clin. Immunol. 2008 122 6 1178 -1184
Kwan A., Manning M.A., Zollars L.K., and Hoyme H.E. Marked variability in the radiographic features of cartilage-hair hypoplasia: Case report and review of the literature Am. J. Med. Genet. A. 2012 158A 11 2911 -2916
Mäkitie O., Ellis L., Durie P.R., Morrison J.A., Sochett E.B., Rommens J.M., and Cole W.G. Skeletal phenotype in patients with Shwachman-Diamond syndrome and mutations in SBDS Clin. Genet. 2004 65 2 101 -112
Mäkitie O. and Kaitila I. Cartilage-hair hypoplasia—Clinical manifestations in 108 Finnish patients Eur. J. Pediatr. 1993 152 3 211 -217
Mäkitie O., Marttinen E., and Kaitila I. Skeletal growth in cartilage-hair hypoplasia. A radiological study of 82 patients Pediatr. Radiol. 1992 22 6 434 -439
Makitie O., Kaitila I., and Savilahti E. Susceptibility to infections and in vitro immune functions in cartilage-hair hypoplasia Eur. J. Pediatr. 1998 157 10 816 -820
Makitie O., Sulisalo T., de la Chapelle A., and Kaitila I. Cartilage-hair hypoplasia J. Med. Genet. 1995 32 1 39 -43
McKusick V.A., Eldridge R., Hostetler J.A., Ruangwit U., and Egeland J.A. Dwarfism in the Amish. II. Cartilage-hair hypoplasia Bull. Johns Hopkins Hosp. 1965 116 285 -326
Notarangelo L.D., Roifman C.M., and Giliani S. Cartilage-hair hypoplasia: Molecular basis and heterogeneity of the immunological phenotype Curr. Opin. Allergy Clin. Immunol. 2008 8 6 534 -539
Ray H.C. and Dorst J.P. Cartilage-hair hypoplasia Progr. Pediatr. Radiol. 1973 4 270
Ridanpää M., van Eenennaam H., Pelin K., Chadwick R., Johnson C., Yuan B., van Venrooij W., Pruijn G., Salmela R., Rockas S., Mäkitie O., Kaitila I., and De La Chapelle A. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia Cell. 2001 104 2 195 -203
Rider N.L., Morton D.H., Puffenberger E., Hendrickson C.L., Robinson D.L., and Strauss K.A. Immunologic and clinical features of 25 Amish patients with RMRP 70 A-->G cartilage hair hypoplasia Clin. Immunol. 2009 131 1 119 -128
Roifman C.M. Antibody deficiency, growth retardation, spondyloepiphyseal dysplasia and retinal dystrophy: A novel syndrome Clin. Genet. 1999 55 2 103 -109
Roifman C.M. and Melamed I. A novel syndrome of combined immunodeficiency, autoimmunity and spondylometaphyseal dysplasia Clin. Genet. 2003 63 6 522 -529
Roifman C.M., Gu Y., and Cohen A. Mutations in the RNA component of RNase mitochondrial RNA processing might cause Omenn syndrome J. Allergy Clin. Immunol. 2006 117 4 897 -903
Sulisalo T., Sistonen P., Hästbacka J., Wadelius C., Mäkitie O., de la Chapelle A., and Kaitila I. Cartilage-hair hypoplasia gene assigned to chromosome 9 by linkage analysis Nat. Genet. 1993 3 4 338 -341
Thiel C.T., Mortier G., Kaitila I., Reis A., and Rauch A. Type and level of RMRP functional impairment predicts phenotype in the cartilage hair hypoplasia–anauxetic dysplasia spectrum Am. J. Hum. Genet. 2007 81 3 519 -529
Van der Burgt I., Haraldsson A., Oosterwijk J.C., van Essen A.J., Weemaes C., and Hamel B. Cartilage hair hypoplasia, metaphyseal chondrodysplasia type McKusick: Description of seven patients and review of the literature Am. J. Med. Genet. 1991 41 3 371 -380
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 3 • September 2015
Pages: 157 - 164
History
Received: 2 June 2015
Accepted: 22 June 2015
Accepted manuscript online: 25 June 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Yoram Faitelson and David Manson. 2015. Cartilage–hair hypoplasia: a spectrum of clinical and radiological findings. LymphoSign Journal.
2(3): 157-164. https://doi.org/10.14785/lpsn-2015-0009
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
