Dendritic cell defects in primary immunodeficiency disorders
Abstract
Dendritic cells (DC) are professional antigen-presenting cells that play a key role in linking the innate and adaptive arms of the immune system. In vitro, DC perform critical functions such as antigen uptake and processing, priming of naïve T cells and production of cytokines to regulate other immune cells. In vivo experimental systems support a central role for DC in inducing protective immune responses but the effect of DC deficiency in existing whole animal models is smaller than would be predicted. Studies of human primary immunodeficiency disorders (PID) have significantly advanced our understanding of the development and function of other immune cells and provide some important information about DC. Although only a small number of rare monogenic PID that cause DC deficiency have been described to date, impaired DC function forms part of the immunophenotype of several PID and is likely to contribute to clinical presentation. This review focuses on what is known so far about the role of DC in PID and what implications this has for basic DC biology.
Introduction
The 2011 Nobel Prize in Physiology or Medicine was awarded to Ralph Steinman for his original discovery of dendritic cells (DC) and subsequent work highlighting their role in immunity. Over the past 4 decades of DC research, these cells have emerged as a heterogenous group of highly specialized innate immune cells that initiate and regulate adaptive immune responses. They are thus predicted to be essential for host protection from pathogens and maintenance of self-tolerance. Although the role of DC has been extensively researched in laboratory models, a clear role for DC in human disease has been harder to clarify.
Abnormal immune responses, both immune deficiency and autoimmunity, are hallmark features of primary immunodeficiency disorders (PID): a group of inherited conditions in which one or more components of the immune system is lacking. Our understanding of the molecular basis of PID has advanced rapidly since the advent of new generation genetic sequencing and now over 300 single gene defects are known to cause separate PID (Al-Herz et al. 2014). Defects of adaptive immune cells, namely T and B lymphocytes, make up the majority of known PID although disorders of innate immunity are increasingly being recognized. In spite of this meteoric advance in PID, DC defects have made little appearance in the field.
This is somewhat surprising given the accepted role for DC in the immune response and presents a conundrum that challenges our understanding of DC biology. Viewed simply, the almost total lack of DC-specific PIDs could indicate either that DC are essential for post-natal viability and therefore DC deficiency is not compatible with life or that DC play a less important, partially redundant, role in whole organism immune responses. However, the explanation may be more complex as there are a number of practical challenges in studying DC that have hampered progress in identifying specific DC deficiencies. Firstly, reliably identifying DC subsets, particularly in tissues, remains problematic as current cell surface markers used for DC identification are generally not are cell specific, but are shared with other haematopoietic cells. Secondly, it is still not known which haematopoietic precursors specifically give rise to DC to enable models of lineage-specific deletion for predication of phenotype. Thirdly, in vitro analysis of DC function has many technical limitations, such as inherent DC activation, that preclude direct translation of results to the in vivo setting.
In the current review, I will briefly outline normal human DC biology and describe the important insights that we have learnt from the field of PID.
Human DC subsets
DC represent a heterogenous group of cells that can be classified based on location, function, surface marker expression, and ontogeny. Over the years, a number of different models of DC ontogeny have been purported and revised as new data emerges. It is currently accepted that DC in humans can broadly be divided into 2 main subsets: myeloid DC (mDC) and plasmacytoid DC (pDC). mDC also known as conventional DC, are the majority subset and function as professional antigen-presenting cells, whereas pDC mainly circulate and have a specialized role in generating Type I Interferon responses important for antiviral responses.
For human DC, most information has been obtained from circulating cells and nomenclature for these blood DC subsets has been agreed upon (Ziegler-Heitbrock et al. 2010). Two circulating populations of mDC can be differentiated based on surface marker expression: a larger CD1c (BDCA-1)+ population and a smaller CD141+ (BDCA-3+) subset (Figure 1). pDC are identified as CD303+ (BDCA-2+) cells. The precursors for these DC subsets are not known in humans, although both granulocyte-macrophage progenitors and multi-lymphoid progenitors, which arise from haematopoietic stem cells, appear to have DC potential (Doulatov et al. 2010).
Figure 1:

In addition to circulating DC, a number of tissue resident DC subsets can be identified. Both CD1c+ and CD141+ DC subsets are found in nonlymphoid tissues, and they can take up antigen, migrate to lymph nodes and prime T cells (Haniffa et al. 2013). The epidermis contains Langerhan cells (LC), a specific DC subset characterized by a high expression of CD1a+. LC perform functions similar to CD1c+ and CD141+ DC but are capable of self-renewal at skin sites, suggesting a separate ontogeny (Igyártó and Kaplan 2013). A fourth subset of CD14+ DC was also thought to exist in peripheral tissues but recent data suggest that these cells are in fact monocyte-derived macrophages rather than bona-fide DC, highlighting the difficulty of subset definition (McGovern et al. 2014).
Broadly speaking, human DC have murine counterparts, although their identification based on surface marker expression differs, which complicates direct comparison (Haniffa et al. 2013).
The roles of DC in host protection and autoimmunity
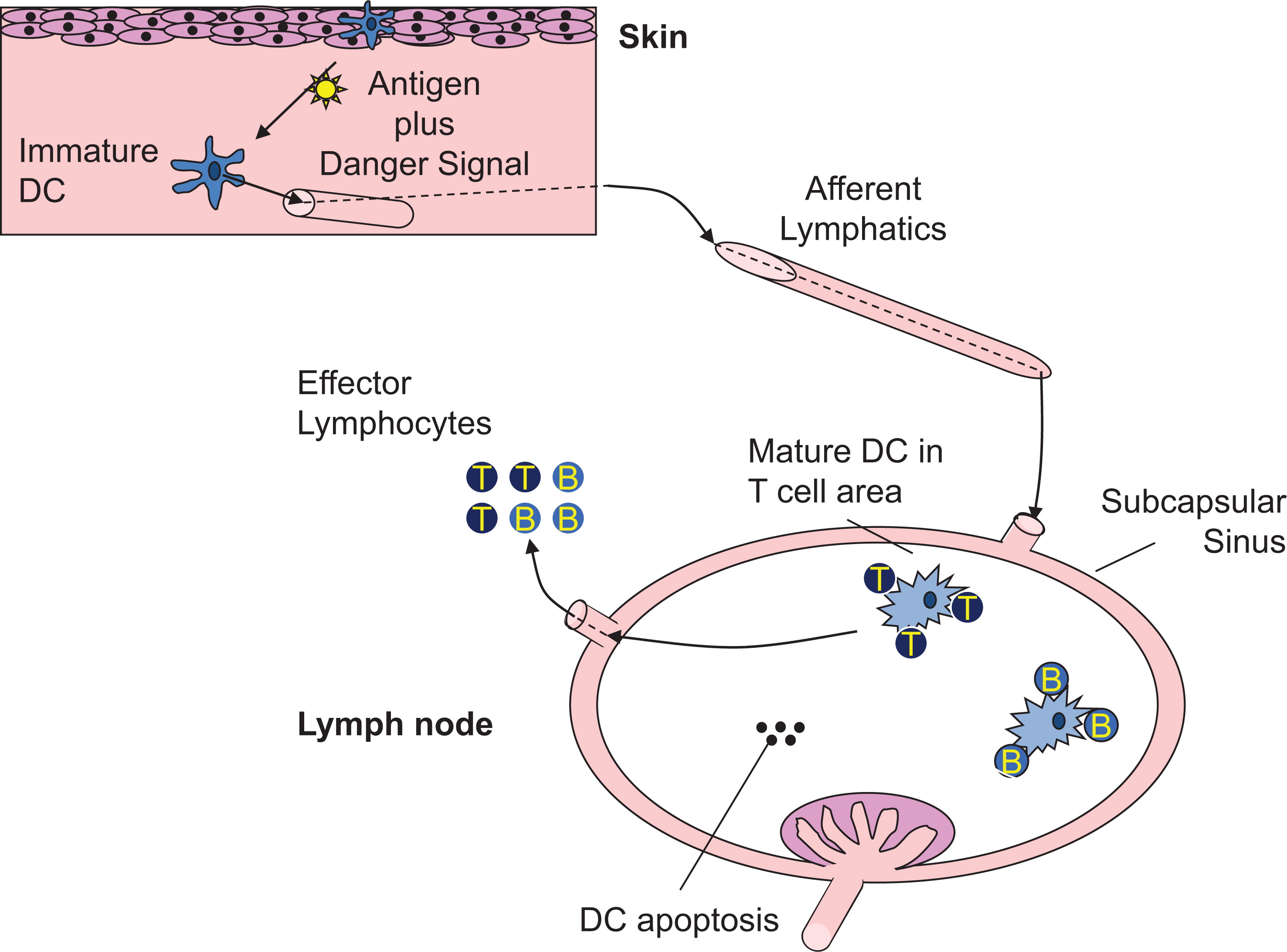
Conventional DC are professional antigen-presenting cells capable of initiating adaptive immune responses and promoting self-tolerance through interaction with T and B cells in secondary lymphoid tissues (Banchereau et al. 2000; Mildner and Jung 2014; Figure 2). In nonlymphoid tissues, mDC reside in an immature state characterized by constitute uptake of exogenous antigen through pinocytosis, endocytosis, and phagocytosis (Inaba et al. 1993; Sallusto et al. 1995). Self and foreign protein antigens, either captured through this environmental surveillance or present in the cytosol, are processed to peptides and loaded onto MHCI or MHCII molecules within specialized intracellular vesicles (Neefjes et al. 2011; Joffre et al. 2012). Delivery of MHC/peptide complexes (pMHC) to the plasma membrane is a key event to enable antigen presentation to CD4+ and CD8+ T cells for T-cell priming and is regulated through DC activation by a variety of stimuli. Maturation stimuli, such as inflammatory cytokines or Toll-like receptor (TLR) ligands, have several additional effects on DC function that promote and regulate antigen presentation. Firstly, DC migration from peripheral to secondary lymphoid tissues is enhanced by DC maturation, facilitating co-location with antigen-specific T cells. Secondly, DC surface expression of co-stimulatory molecules such as CD80 and CD86, necessary for optimal T-cell activation, is up regulated, and thirdly, activation results in DC cytokine produc-tion that drives T-cell effector differentiation. The net effect is potent antigen presentation, a prerequisite for adaptive immunity and effective host protection. How DC induce a strong antipathogen immune response without simultaneously causing T-cell activation to self-antigens is an area of significant interest. It is known that in the absence of maturation, DC can induce regulatory T cells and T-cell anergy to promote self-tolerance, but other mechanisms by which DC confer immune tolerance remain to be fully elucidated (Ganguly et al. 2013).
Figure 2:
In contrast with mDC, pDC do not appear to operate as professional antigen-presenting cells. Instead they are equipped to secrete large amounts of Type I interferons (IFN) in response to ligation of TLR 7 or 9, suggesting a role in viral clearance. Their role in the pathogenesis of autoimmune diseases such as systemic lupus erythematosus, where Type I IFN are thought to be contributory, remains a prominent research focus with potential importance for PID (Ganguly et al. 2013).
DC deficiency in mice: what do we learn?
Based on the unique ability of DC to prime T-cell responses and their role in tolerance, it might reasonably be expected that infection and autoimmunity would characterize DC deficiency states. Mouse models were initially developed to address this question. In the first of these, the diptheria toxin receptor was expressed under the control of the CD11c promoter (Jung et al. 2002), permitting deletion of all CD11c+ cells following injection of diphtheria toxin in postnatal mice. This effectively deletes mDC and pDC, inducing a DC deficiency state that can be experimentally manipulated. There are, however, several important limitations to the system that were recently reviewed in detail (van Blijswijk et al. 2013). Firstly, specificity is not complete, as CD11c is expressed on some other immune subsets so that outcomes may be affected by cells other than DC. Secondly, the duration of depletion is limited by diphtheria toxicity, so that long-term experiments are not feasible. Thirdly, DC ablation in this model is associated with significant neutrophilia and monocytosis that confounds assessment of the innate immune response. In spite of these limitations, these mice have been used to examine the role of DC in a number of infectious models. The summary of these data shows that, at least using this model, DC are required for optimal host defence against specific pathogens (such as toxoplasma). A broad infection susceptibility has not been observed, but this may reflect increased neutrophil and monocyte function that confer improved bacterial defence following depletion of DC (Autenrieth et al. 2012). With regard to tolerance, DC depletion using a DTR mouse model has been reported to be associated with fatal spontaneous autoimmunity, suggesting that human DC deficiency syndromes should also be associated with autoimmunity (Ohnmacht et al. 2009).
Defects of DC development
Many monogenic PIDs disrupt the development of T and B cells, but until recently no genetic disorders were known to selectively impair DC development and thereby shed further light on the impact of DC deficiency. Identification of interferon response factor 8 (IRF8) deficiency has provided the best example of a predominantly DC-mediated PID, and although only a few cases have been described in a single report, this condition provides important insight into the role of human DC for host protection (Hambleton et al. 2011). Autosomal recessive IRF8 deficiency leads to developmental defects of DC and monocytes, with a complete absence of monocytes, circulating mDC and pDC, and a paucity of tissue-resident dermal DC, suggesting a common progenitor for these subsets. In contrast, Langerhan DC are preserved, in keeping with a distinct ontogeny. DC and monocyte deficiency in the single case described, caused a remarkably narrow spectrum of infection, limited to disseminated infection with vaccine strain Bacillus Calmette–Guérin (BCG), severe respiratory viruses, and candidiasis. This may be explained, in part, by elevated circulating neutrophils associated with high levels of serum Flt-3 ligand and myeloid hyperplasia in bone marrow and BCG infected lymph nodes that could confer bacterial protection, reminiscent of murine models of DC depletion (Birnberg et al. 2008; Autenrieth et al. 2012).
In the same study (Hambleton et al. 2011), autosomal dominant IRF8 deficiency was also described in 2 unrelated adults with heterozygous loss of function mutations in IRF8 (IRF8T80A). This single mutation appears to have has a more selective effect as only the subset of CD1c+ mDC was reduced resulting in isolated recurrent BCG disease, reinforcing the concept that DC are nonredundant for protection against mycobacteria. Reduced CD1c+ mDC was associated with a reduced production of IL-12 by peripheral blood mononuclear cells (PBMC) in response to TLR 7/8 stimulation, providing a plausible explanation for mycobacterial susceptibility in these patients.
A second transcription factor, GATA2, has since been identified as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency (also known as DCML deficiency or Monomac) (Bigley et al. 2011; Dickinson et al. 2011; Hsu et al. 2011). Compared with IRF8 deficiency, loss-of-function mutations in GATA2 have a broader effect on cell development. Both lymphoid and myeloid precursor cells are reduced in number, resulting in low levels of NK and B cells in addition to severe moncytopoenia and an almost total absence of circulating and tissue DC. As in IRF8 deficiency, Langerhans cells are preserved. An intriguing feature of GATA2 deficiency is its late onset; patients often do not present until the third decade of life, although monocytopoenia may have been present for many years beforehand. This suggests exhaustion of precursors over time rather than an early developmental defect as seen in IRF8, but this phenotype remains poorly understood. Similar to autosomal recessive IRF8 deficiency, GATA2 defects confer a relatively selective infection susceptibility to nontuberculous mycobacteria, opportunistic fungi, and some viruses, particularly human papilloma and herpes viruses. The condition has a number of apparently noninfectious life-threatening complications that do not relate directly to immunodeficiency, the most serious of which are alveolar proteinosis, myelodysplasia, and myeloid leukaemia.
Together, IRF8 and GATA-2 deficiencies suggest that the loss of DC has a selective effect on host protection with a particular importance for immunity against mycobacteria. Further descriptions of IRF8 deficiency and identification of other novel DC-specific PID are required to clarify DC-dependent immune outcomes. Perhaps an important parameter to measure in DC deficiency is not acute infection but successful induction of T-cell memory for specific organisms, which would be predicted to depend on DC-mediated antigen presentation. This is supported by RNA-expression profiling of AR IRF8-deficiency, which has revealed a depletion of gene signatures for memory CD4+ and CD8+ T-cells, consistent with a defect of antigen presentation (Salem et al. 2014). Finally, neither the IRF8 or GATA-2 deficiencies have been described to be associated with autoimmunity and long term follow-up, plus description of more cases will be informative.
PID that impair DC function
In contrast with DC deficiency states, several combined immunodeficiencies have been described in which DC function is impaired. Unlike NK-cell deficiencies, which can represent a major innate component of a combined immune defect (Orange 2013), the contribution of DC dysfunction to the clinical spectrum of combined immunodeficiency syndromes is unclear. Data currently available for these conditions, therefore, mainly provide information about the role of specific genes and proteins for aspects of DC function and highlight the importance that PID have played in understanding normal immune cell biology.
Wiskott–Aldrich syndrome
Probably the best studied PID with regard to DC is Wiskott–Aldrich syndrome (WAS): an X-linked condition that results from loss of function mutations in the WAS gene (Burns et al. 2004). WAS protein (WASp) is a cytoskeletal regulator that is expressed in all haematopoietic cells and is required for many cellular functions such as phagocytosis, cell migration, and immune synapse formation (Thrasher and Burns 2010). Absence of WASp function results in a combined immunodeficiency that impairs the function of T, B, and NK cells as well as myeloid lineage cells. The clinical phenotype is broad as a result: characterized by recurrent bacterial, viral and fungal infections, autoimmunity, bleeding (as a result of platelet defects), severe eczema, and an increased risk of haematological malignancies.
Like other immune cells, DC rely on WASp for f-actin assembly (Binks et al. 1998). It is therefore not that surprising that cytoskeletal-associated functions in DC are impaired by WASp deficiency. Initial studies focused on migration and demonstrated that mDC from patients (and mice) with WAS fail to form podosomes, F-actin rich structures that link the cytoskeleton to membrane integrins (Binks et al. 1998; Burns et al. 2001; Olivier et al. 2005). Cell adhesion and migration in vitro are impaired and the in vivo consequence is a failure of WASp-deficient DC to migrate to lymph nodes for antigen presentation to T cells (Burns et al. 2001; de Noronha et al. 2005; Bouma et al. 2007). At least in murine models, migration defects in WASp-deficient DC impacts initiation of CD4+ and CD8+ adaptive T-cell responses that would be predicted to be biologically significant (Bouma et al. 2007; Pulecio et al. 2008). Antigen presentation is further impacted in DC that lack WASp by defective antigen uptake via phagocytosis and impaired formation of an immune synapse with T cells (Westerberg et al. 2003; Bouma et al. 2007; Pulecio et al. 2008). Although most studies have focused on the cross-talk between DC and T cells, cell–cell interaction also occurs between DC and other cell types, and it has recently been shown that NK cell activation is impaired if DC lack WASp (Catucci et al. 2014). This has potential implications for malignancy in WAS, as NK cells activated by WASp-deficient DC were unable in mouse models to control tumour spread.
The role of pDC in the pathogenesis of WAS has only recently received attention, and to date 2 separate studies have shown that WASp deficiency impacts pDC function, with somewhat different conclusions. In humans, WASp deficiency was reported to result in lower levels of pDC in peripheral blood (Prete et al. 2013). However, instead of resulting in low levels of Type I IFN, elevated baseline and stimulated levels of IFN-α were observed. Prete et al. (2013) demonstrated, in a very elegant study, that WASp-deficient pDC are intrinsically hyper-responsive to TLR9 ligands, such as double-stranded DNA because WASp is required for trafficking of TLR9 ligands into the endo-lysosomal compartment in pDC and regulation of TLR9-induced IFN-α responses. This could represent one mechanism for autoimmunity in WAS, where autoantibody/self DNA immune complexes are found at higher levels in the serum and act as a ligand for TLR9 (Recher et al. 2012; Prete et al. 2013). A second study also reported low numbers of Type I IFN producing pDC and mDC in mice (Lang et al. 2013), which was associated with an increased susceptibility to viral infection with lymphocytic choriomeningitis virus (LCMV). Although, at first, this appears incompatible with the observation that WASp deficiency enhances IFN-α responses, murine pDC are chronically activated in vivo, presumably by circulating immune complexes, which then results in exhaustion following additional stimulation (Prete et al. 2013). Thus it is feasible on the one hand, to have elevated basal levels of IFN-α causing autoimmunity in WAS and on the other hand to have impaired IFN-α up-regulation during infection resulting in viral susceptibility. These data suggest that viral susceptibility in patents with WAS in part relates to lower Type I IFN responses, which could have therapeutic implications.
DOCK8 deficiency
Like WASp, DOCK8 normally functions to regulate the actin cytoskeleton through activation of the Rho-family GTPase Cdc42. Patients with DOCK8 deficiency have a clinical picture that resembles WAS with a combined immunodeficiency and broad susceptibility to bacterial and viral infections associated with atopic dermatitis (Engelhardt et al. 2009; Zhang et al. 2009). At a cellular level, DOCK8 appears to have a more limited role in DC f-actin assembly than WASp, reflected in a less severe effect of DOCK8 deficiency on mDC actin structures and migration. DOCK8 mDC are capable of assembling podosomes (personal communication G. Bouma and A.J. Thrasher) and can migrate in 2-D experimental systems (Harada et al. 2012). However, the ability of DOCK8 deficient DC to change shape and migrate in 3-D environments and in vivo is significantly impaired and results in reduced T-cell priming (Harada et al. 2012), much like the findings seen in WASp-deficient mice. Patients with DOCK8 deficiency also demonstrate low numbers of pDC in peripheral blood. This is associated with dramatically reduced levels of IFN-α production in response to stimulation that was not explained fully by pDC numbers, and likely reflects an additional intrinsic pDC functional defects that remains to be fully clarified (Al-Zahrani et al. 2014; Keles et al. 2014). In keeping with a central role for pDC in viral susceptibility in DOCK8 deficient patients, treatment with exogenous IFN-α resulted in resolution of herpes and human papilloma virus infections in 2 patients (Al-Zahrani et al. 2014; Keles et al. 2014). However, as T and B cells are also defective in DOCK8 deficiency, it is difficult to tease out with confidence the relative contribution of DC to host protection.
Hyper IgM syndromes
Type 1 and Type 3 hyper IgM syndrome are caused by loss of function mutations in CD40 and its ligand CD154 (also known as CD40L). It is well known that interaction between CD40 expressed on B cells and CD40L on T cells is critical for cross-talk between these cell types, and that the absence of either molecule prevents immunoglobulin class switching, resulting in antibody deficiency. The clinical spectrum of disease, however, is broader than would be suggested by a humoral defect and includes susceptibility to opportunistic infection characteristic of T-cell deficiencies. DC also express CD40, and ligation by CD154 provides a maturation stimulus. Although CD40 expression by DC does not appear to be required for mDC or pDC development, CD40 deficient monocyte-derived DC are unable to up-regulate MHCII and co-stimulatory molecules such as CD80 and CD86 when activated by CD40L (Fontana et al. 2003). This is accompanied by reduced IL-12 and elevated IL-10 release and impaired induction of T-cell activation and proliferation. Curiously, very similar effects on DC function are found in patients with CD40L deficiency, where monocyte-derived DC also demonstrate lower expression of MHCII and co-stimulatory molecules and imbalanced IL-12/IL-10 release that can be rescued by co-culture with soluble CD40L but not with other physiological stimuli (Cabral-Marques et al. 2012). DC from patients with CD40L deficiency are capable of inducing normal autologous T-cell proliferation, but with perturbed T-cell cytokine production towards a TH2 phenotype. This suggests that failure to provide CD40--CD40L signaling in DC impacts DC induction of T-cell responses regardless of which molecule is lacking, and this might represent one mechanism for T-cell dysfunction in these syndromes.
Hyper IgE syndrome
STAT3 is a widely expressed molecule that is commonly utilized to transduce signals by multiple cytokines at the cell surface. Loss of function mutations in STAT3 account for the majority of cases of hyper IgE syndrome, a complex immunodeficiency characterized by staphylococcal abscesses, chest infections, candidiasis along with eczema, elevated IgE, bone abnormalities, and dysmorphic features. Multiple immune cells rely on STAT3 for normal function, of which DC are at least one such cell type. Although several cytokine receptors in DC signal using STAT3, it has been shown that defects in the IL-10 signaling pathway in mDC result in a failure to regulate DC activation (Giacomelli et al. 2011; Saito et al. 2011). Both monocyte-derived and ex-vivo primary mDC from STAT3-deficient patients fail to down-regulate MHCII, co-stimulatory molecules, and inflammatory cytokines in response to IL-10. This results in a failure to develop “tolerogenic DC” and a consequent incapacity to induce T regulatory cells that may contribute to immune dysregulation and inflammatory complications associated with STAT3 deficiency.
Common variable immune deficiency (CVID)
Although there have been a number of publications examining DC function in CVID there remains controversy about whether DC defects are intrinsic or secondary to environmental factors in this condition. Unlike the monogenic PIDs discussed so far, CVID represents a heterogenous group of disorders that result in a defined antibody deficiency (Jolles 2013). A very small number of CVID cases have an identified genetic basis and for the most part the molecular pathogenesis remains unknown. Although DC studies published to date have included CVID patients in general, it seems most probable that if DC contribute to the pathogenesis of CVID, this would only be true for a subset of patients and not a general mechanism.
The most consistent finding among CVID studies is a reduction in the numbers of circulating mDC and pDC (Viallard et al. 2005; Martinez-Pomar et al. 2006; Paquin-Proulx et al. 2013; Taraldsrud et al. 2014). The reason for this is not clear although, at least for pDC, it does not appear to be due to reduced bone marrow production or due to sequestration in peripheral tissues (Paquin-Proulx et al. 2013). Peripheral destruction is possible and would be supported by the findings that lower pDC levels correlate with the presence of splenomegaly and that mDC numbers can be restored by IVIG treatment (Paquin-Proulx et al. 2013; Taraldsrud et al. 2014), as is seen for autoimmune cytopoenias such as thrombocytopoenia. Whether there is an intrinsic dysfunction of mDC, in addition to low numbers, remains to be determined. Initial studies, using in vitro testing of monocyte-derived DC, reported defects of DC maturation and subsequent T-cell activation in vitro (Bayry et al. 2004; Scott-Taylor et al. 2004, 2006). Although this could be a plausible mechanism for immunodeficiency, it has not been verified in vivo and, in fact, maturation markers measured on circulating mDC ex vivo are not reduced but in fact increased (Bayry 2004; Paquin-Proulx et al. 2013).
More recently, considerable interest has been focused on the question of whether CVID pDC contribute to disease pathogenesis. Initial stimulation of ex-vivo sorted pDC demonstrated impaired IFN-α responses to TLR-9 ligands (Cunningham-Rundles et al. 2006) and follow-up studies reported that pDC responses to TLR-7 ligation was similarly defective in CVID (Yu et al. 2009, 2012). This appeared to provide a sensible link between DC defects and B cell dysfunction, as IFN-α is known to augment TLR-induced B-cell activation and maturation (Bekeredjian-Ding et al. 2005). In support of the hypothesis that pDC-released IFN-α is a disease mechanism in CVID, defective TLR7-induced class switching in CVID B cells could be partially rescued by the addition of exogenous IFN-α (Yu et al. 2009, 2012). However, a separate group concluded that there is no intrinsic defect in IFN-α production by pDC in CVID, but that reduced numbers of the pDC population was responsible for reduced IFN-α production by TLR7-stimulated PBMC in their study (Taraldsrud et al. 2014). It remains possible that low numbers of pDC are sufficient to impair IFN-α responses and contribute to B-cell dysfunction in CVID, and this is an area that warrants further investigation.
Conclusions
Studies of DC involvement in PID have provided some important insights into basic DC biology. Although only a handful of patients with specific DC deficiency states have been described, so far they have not been characterized by broad infection susceptibility. Rather they suggest that DC play a crucial role in host defence against specific pathogens, notably mycobacteria. Identification of additional cases of DC deficiency is required to substantiate this and clarify the importance of DC for generation of successful T-cell memory. Autoimmunity has thus far not been a prominent feature of DC deficiency, although it is commonly associated with combined immunodeficiency states where DC function is impaired. Analysis of DC function in monogenic PID has been particularly valuable for identifying molecules critical for normal DC biology (summarized in Figure 3), but the challenge for the future is to tease out the specific contribution of DC to the overall immune dysfunction in these conditions. Improved understanding of the role of DC in specific PID has particular importance for determining whether bone marrow transplantation and gene therapy treatment protocols should aim to fully correct this cellular compartment.
Figure 3:

Acknowledgements
The author is supported by the Higher Education Funding Council for England.
REFERENCES
Al-Herz W., Bousfiha A., Casanova J.L., Chatila T., Conley M.E., Cunningham-Rundles C., Etzioni A., Franco J.L., Gaspar H.B., Holland S.M., Klein C., Nonoyama S., Ochs H.D., Oksenhendler E., Picard C., Puck J.M., Sullivan K., and Tang M.L. Corrigendum: Primary immunodeficiency diseases: An update on the classification from the International Union of Immunological Societies Expert Committee for primary immunodeficiency Front. Immunol 2014 5 460
Al-Zahrani D., Raddadi A., Massaad M., Keles S., Jabara H.H., Chatila T.A., and Geha R. Successful interferon-alpha 2b therapy for unremitting warts in a patient with DOCK8 deficiency Clin. Immunol 2014 153 104 -108
Autenrieth S.E., Warnke P., Wabnitz G.H., Lucero Estrada C., Pasquevich K.A., Drechsler D., Günter M., Hochweller K., Novakovic A., Beer-Hammer S., Samstag Y., Hämmerling G.J., Garbi N., and Autenrieth I.B. Depletion of dendritic cells enhances innate anti-bacterial host defense through modulation of phagocyte homeostasis PLoS Pathog 2012 8 e1002552
Banchereau J., Briere F., Caux C., Davoust J., Lebecque S., Liu Y.J., Pulendran B., and Palucka K. Immunobiology of dendritic cells Annu. Rev. Immunol 2000 18 767 -811
Bayry J., Lacroix-Desmazes S., Kazatchkine M.D., Galicier L., Lepelletier Y., Webster D., Lévy Y., Eibl M.M., Oksenhendler E., Hermine O., and Kaveri S.V. Common variable immunodeficiency is associated with defective functions of dendritic cells Blood 2004 104 2441 -2443
Bekeredjian-Ding I.B., Wagner M., Hornung V., Giese T., Schnurr M., Endres S., and Hartmann G. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN J. Immunol 2005 174 4043 -4050
Bigley V., Haniffa M., Doulatov S., Wang X.N., Dickinson R., McGovern N., Jardine L., Pagan S., Dimmick I., Chua I., Wallis J., Lordan J., Morgan C., Kumararatne D.S., Doffinger R., van der Burg M., van Dongen J., Cant A., Dick J.E., Hambleton S., and Collin M. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency J. Exp. Med 2011 208 227 -234
Binks M., Jones G.E., Brickell P.M., Kinnon C., Katz D.R., and Thrasher A.J. Intrinsic dendritic cell abnormalities in Wiskott-Aldrich syndrome Eur. J. Immunol 1998 28 3259 -3267
Birnberg T., Bar-On L., Sapoznikov A., Caton M.L., Cervantes-Barragán L., Makia D., Krauthgamer R., Brenner O., Ludewig B., Brockschnieder D., Riethmacher D., Reizis B., and Jung S. Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome Immunity 2008 29 986 -997
Bouma G., Burns S., and Thrasher A.J. Impaired T-cell priming in vivo resulting from dysfunction of WASp-deficient dendritic cells Blood 2007 110 4278 -4284
Burns S., Cory G.O., Vainchenker W., and Thrasher A.J. Mechanisms of WASp-mediated hematologic and immunologic disease Blood 2004 104 3454 -3462
Burns S., Thrasher A.J., Blundell M.P., Machesky L., and Jones G.E. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation Blood 2001 98 1142 -1149
Cabral-Marques O., Arslanian C., Ramos R.N., Morato M., Schimke L., Soeiro Pereira P.V., Jancar S., Ferreira J.F., Weber C.W., Kuntze G., Rosario-Filho. N.A., Costa Carvalho B.T., Bergami-Santos P.C., Hackett M.J., Ochs H.D., Torgerson T.R., Barbuto J.A., and Condino-Neto A. Dendritic cells from X-linked hyper-IgM patients present impaired responses to Candida albicans and Paracoccidioides brasiliensis J. Allergy Clin. Immunol 2012 129 778 -786
Catucci M., Zanoni I., Draghici E., Bosticardo M., Castiello M.C., Venturini M., Cesana D., Montini E., Ponzoni M., Granucci F., and Villa A. Wiskott-Aldrich syndrome protein deficiency in natural killer and dendritic cells affects antitumor immunity Eur. J. Immunol 2014 44 1039 -1045
Cunningham-Rundles C., Radigan L., Knight A.K., Zhang L., Bauer L., and Nakazawa A. TLR9 activation is defective in common variable immune deficiency J. Immunol 2006 176 1978 -1987
de Noronha S., Hardy S., Sinclair J., Blundell M.P., Strid J., Schulz O., Zwirner J., Jones G.E., Katz D.R., Kinnon C., and Thrasher A.J. Impaired dendritic-cell homing in vivo in the absence of Wiskott-Aldrich syndrome protein Blood 2005 105 1590 -1597
Dickinson R.E., Griffin H., Bigley V., Reynard L.N., Hussain R., Haniffa M., Lakey J.H., Rahman T., Wang X.N., McGovern N., Pagan S., Cookson S., McDonald D., Chua I., Wallis J., Cant A., Wright M., Keavney B., Chinnery P.F., Loughlin J., Hambleton S., Santibanez-Koref M., and Collin M. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency Blood 2011 118 2656 -2658
Doulatov S., Notta F., Eppert K., Nguyen L.T., Ohashi P.S., and Dick J.E. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development Nat. Immunol 2010 11 585 -593
Engelhardt K.R., McGhee S., Winkler S., Sassi A., Woellner C., Lopez-Herrera G., Chen A., Kim H.S., Lloret M.G., Schulze I., Ehl S., Thiel J., Pfeifer D., Veelken H., Niehues T., Siepermann K., Weinspach S., Reisli I., Keles S., Genel F., Kutukculer N., Camcıoğlu Y., Somer A., Karakoc-Aydiner E., Barlan I., Gennery A., Metin A., Degerliyurt A., Pietrogrande M.C., Yeganeh M., Baz Z., Al-Tamemi S., Klein C., Puck J.M., Holland S.M., McCabe E.R., Grimbacher B., and Chatila T.A. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome J. Allergy. ClinImmunol 2009 124 1289 -1302
Fontana S., Moratto D., Mangal S., De Francesco M., Vermi W., Ferrari S., Facchetti F., Kutukculer N., Fiorini C., Duse M., Das P.K., Notarangelo L.D., Plebani A., and Badolato R. Functional defects of dendritic cells in patients with CD40 deficiency Blood 2003 102 4099 -4106
Ganguly D., Haak S., Sisirak V., and Reizis B. The role of dendritic cells in autoimmunity Nat. Rev. Immunol 2013 13 566 -577
Giacomelli M., Tamassia N., Moratto D., Bertolini P., Ricci G., Bertulli C., Plebani A., Cassatella M., Bazzoni F., and Badolato R. SH2-domain mutations in STAT3 in hyper-IgE syndrome patients result in impairment of IL-10 function Eur. J. Immunol 2011 41 3075 -3084
Hambleton S., Salem S., Bustamante J., Bigley V., Boisson-Dupuis S., Azevedo J., Fortin A., Haniffa M., Ceron-Gutierrez L., Bacon C.M., Menon G., Trouillet C., McDonald D., Carey P., Ginhoux F., Alsina L., Zumwalt T.J., Kong X.F., Kumararatne D., Butler K., Hubeau M., Feinberg J., Al-Muhsen S., Cant A., Abel L., Chaussabel D., Doffinger R., Talesnik E., Grumach A., Duarte A., Abarca K., Moraes-Vasconcelos D., Burk D., Berghuis A., Geissmann F., Collin M., Casanova J.L., and Gros P. IRF8 mutations and human dendritic-cell immunodeficiency N. Engl. J. Med 2011 365 127 -138
Haniffa M., Collin M., and Ginhoux F. Ontogeny and functional specialization of dendritic cells in human and mouse Adv. Immunol 2013 120 1 -49
Harada Y., Tanaka Y., Terasawa M., Pieczyk M., Habiro K., Katakai T., Hanawa-Suetsugu K., Kukimoto-Niino M., Nishizaki T., Shirouzu M., Duan X., Uruno T., Nishikimi A., Sanematsu F., Yokoyama S., Stein J.V., Kinashi T., and Fukui Y. DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses Blood 2012 119 4451 -4461
Hsu A.P., Sampaio E.P., Khan J., Calvo K.R., Lemieux J.E., Patel S.Y., Frucht D.M., Vinh D.C., Auth R.D., Freeman A.F., Olivier K.N., Uzel G., Zerbe C.S., Spalding C., Pittaluga S., Raffeld M., Kuhns D.B., Ding L., Paulson M.L., Marciano B.E., Gea-Banacloche J.C., Orange J.S., Cuellar-Rodriguez J., Hickstein D.D., and Holland S.M. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome Blood 2011 118 2653 -2655
Igyártó B.Z. and Kaplan D.H. Antigen presentation by Langerhans cells Curr. Opin. Immunol 2013 25 115 -119
Inaba K., Inaba M., Naito M., and Steinman R.M. Dendritic cell progenitors phagocytose particulates, including bacillus Calmette-Guerin organisms, and sensitize mice to mycobacterial antigens in vivo J. Exp. Med 1993 178 479 -488
Joffre O.P., Segura E., Savina A., and Amigorena S. Cross-presentation by dendritic cells Nat. Rev. Immunol 2012 12 557 -569
Jolles S. The variable in common variable immunodeficiency: A disease of complex phenotypes J. Allergy Clin. Immunol Pract 2013 1 545 -556
Jung S., Unutmaz D., Wong P., Sano G., De los Santos K., Sparwasser T., Wu S., Vuthoori S., Ko K., Zavala F., Pamer E.G., Littman D.R., and Lang R.A. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens Immunity 2002 17 211 -220
Keles S., Jabara H.H., Reisli I., McDonald D.R., Barlan I., Hanna-Wakim R., Dbaibo G., Lefranc G., Al-Herz W., Geha R.S., and Chatila T.A. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: Rescue of severe herpetic infections with IFN-alpha 2b therapy J. Allergy Clin. Immunol 2014 133 1753 -1755
Lang P.A., Shaabani N., Borkens S., Honke N., Scheu S., Booth S., Brenner D., Meryk A., Barthuber C., Recher M., Mak T.W., Ohashi P.S., Häussinger D., Griffiths G.M., Thrasher A.J., Bouma G., and Lang K.S. Reduced type I interferon production by dendritic cells and weakened antiviral immunity in patients with Wiskott-Aldrich syndrome protein deficiency J. Allergy Clin. Immunol 2013 131 815 -824
Martinez-Pomar N., Raga S., Ferrer J., Pons J., Munoz-Saa I., Julia M.R., de Gracia J., and Matamoros N. Elevated serum interleukin (IL)-12p40 levels in common variable immunodeficiency disease and decreased peripheral blood dendritic cells: Analysis of IL-12p40 and interferon-gamma gene Clin. Exp. Immunol 2006 144 233 -238
McGovern N., Schlitzer A., Gunawan M., Jardine L., Shin A., Poyner E., Green K., Dickinson R., Wang X.N., Low D., Best K., Covins S., Milne P., Pagan S., Aljefri K., Windebank M., Miranda-Saavedra D., Larbi A., Wasan P.S., Duan K., Poidinger M., Bigley V., Ginhoux F., Collin M., and Haniffa M. Human dermal CD14+ cells are a transient population of monocyte-derived macrophages Immunity 2014 41 465 -477
Mildner A. and Jung S. Development and function of dendritic cell subsets Immunity 2014 40 642 -656
Neefjes J., Jongsma M.L., Paul P., and Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation Nat. Rev. Immunol 2011 11 823 -836
Ohnmacht C., Pullner A., King S.B., Drexler I., Meier S., Brocker T., and Voehringer D. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity J. Exp. Med 2009 206 549 -559
Olivier A., Jeanson-Leh L., Bouma G., Compagno D., Blondeau J., Seye K., Charrier S., Burns S., Thrasher A.J., Danos O., Vainchenker W., and Galy A. A partial down-regulation of WASP is sufficient to inhibit podosome formation in dendritic cells Mol. Ther 2006 13 729 -737
Orange J.S. Natural killer cell deficiency J. Allergy Clin. Immunol 2013 132 515 -525
Paquin-Proulx D., Santos B.A., Carvalho K.I., Toledo-Barros M., Barreto de Oliveira A.K., Kokron C.M., Kalil J., Moll M., Kallas E.G., and Sandberg J.K. IVIg immune reconstitution treatment alleviates the state of persistent immune activation and suppressed CD4 T cell counts in CVID PLoS One 2013 8 e75199
Prete F., Catucci M., Labrada M., Gobessi S., Castiello M.C., Bonomi E., Aiuti A., Vermi W., Cancrini C., Metin A., Hambleton S., Bredius R., Notarangelo L.D., van der Burg M., Kalinke U., Villa A., and Benvenuti F. Wiskott-Aldrich syndrome protein-mediated actin dynamics control type-I interferon production in plasmacytoid dendritic cells J. Exp. Med 2013 210 355 -374
Pulecio J., Tagliani E., Scholer A., Prete F., Fetler L., Burrone O.R., and Benvenuti F. Expression of Wiskott-Aldrich syndrome protein in dendritic cells regulates synapse formation and activation of naive CD8+ T cells J. Immunol 2008 181 1135 -1142
Recher M., Burns S.O., de la Fuente M.A., Volpi S., Dahlberg C., Walter J.E., Moffitt K., Mathew D., Honke N., Lang P.A., Patrizi L., Falet H., Keszei M., Mizui M., Csizmadia E., Candotti F., Nadeau K., Bouma G., Delmonte O.M., Frugoni F., Fomin A.B., Buchbinder D., Lundequist E.M., Massaad M.J., Tsokos G.C., Hartwig J., Manis J., Terhorst C., Geha R.S., Snapper S., Lang K.S., Malley R., Westerberg L., Thrasher A.J., and Notarangelo L.D. B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein (WASp) causes severe abnormalities of the peripheral B-cell compartment in mice Blood 2012 119 2819 -2828
Saito M., Nagasawa M., Takada H., Hara T., Tsuchiya S., Agematsu K., Yamada M., Kawamura N., Ariga T., Tsuge I., Nonoyama S., Karasuyama H., and Minegishi Y. Defective IL-10 signaling in hyper-IgE syndrome results in impaired generation of tolerogenic dendritic cells and induced regulatory T cells J. Exp. Med 2011 208 235 -249
Salem S., Langlais D., Lefebvre F., Bourque G., Bigley V., Haniffa M., Casanova J.L., Burk D., Berghuis A., Butler K.M., Leahy T.R., Hambleton S., and Gros P. Functional characterization of the human dendritic cell immunodeficiency associated with the IRF8K108E mutation Blood 2014 124 1894 -1904
Sallusto F., Cella M., Danieli C., and Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products J. Exp. Med 1995 182 389 -400
Scott-Taylor T.H., Green M.R., Eren E., and Webster A.D. Monocyte derived dendritic cell responses in common variable immunodeficiency Clin. Exp. Immunol 2004 138 484 -490
Scott-Taylor T.H., Green M.R., Raeiszadeh M., Workman S., and Webster A.D. Defective maturation of dendritic cells in common variable immunodeficiency Clin. Exp. Immunol 2006 145 420 -427
Taraldsrud E., Fevang B., Aukrust P., Beiske K.H., Fløisand Y., Frøland S., Rollag H., and Olweus J. Common variable immunodeficiency revisited: Normal generation of naturally occurring dendritic cells that respond to Toll-like receptors 7 and 9 Clin. Exp. Immunol 2014 75 439 -448
Thrasher A.J. and Burns S.O. WASP: A key immunological multitasker Nat. Rev. Immunol 2010 10 182 -192
van Blijswijk J., Schraml B.U., and Reis e Sousa C. Advantages and limitations of mouse models to deplete dendritic cells Eur. J .Immunol 2013 43 22 -26
Viallard J.F., Camou F., Andre M., Liferman F., Moreau J.F., Pellegrin J.L., and Blanco P. Altered dendritic cell distribution in patients with common variable immunodeficiency Arthritis Res. Ther 2005 7 R1052 -R1055
Westerberg L., Wallin R.P., Greicius G., Ljunggren H.G., and Severinson E. Efficient antigen presentation of soluble, but not particulate, antigen in the absence of Wiskott-Aldrich syndrome protein Immunology 2003 109 384 -391
Yu J.E., Knight A.K., Radigan L., Marron T.U., Zhang L., Sanchez-Ramón S., and Cunningham-Rundles C. Toll-like receptor 7 and 9 defects in common variable immunodeficiency J. Allergy Clin. Immunol 2009 124 349 -356, 356
Yu J.E., Zhang L., Radigan L., Sanchez-Ramon S., and Cunningham-Rundles C. TLR-mediated B cell defects and IFN-α in common variable immunodeficiency J. Clin. Immunol 2012 32 50 -60
Zhang Q., Davis J.C., Lamborn I.T., Freeman A.F., Jing H., Favreau A.J., Matthews H.F., Davis J., Turner M.L., Uzel G., Holland S.M., and Su H.C. Combined immunodeficiency associated with DOCK8 Mutations N. Engl. J .Med 2009 361 21 2046 -2055
Ziegler-Heitbrock L., Ancuta P., Crowe S., Dalod M., Grau V., Hart D.N., Leenen P.J., Liu Y.J., MacPherson G., Randolph G.J., Scherberich J., Schmitz J., Shortman K., Sozzani S., Strobl H., Zembala M., Austyn J.M., and Lutz M.B. Nomenclature of monocytes and dendritic cells in blood Blood 2010 116 e74 -e80
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 1 • March 2016
Pages: 1 - 12
History
Received: 19 June 2015
Accepted: 11 August 2015
Accepted manuscript online: 11 August 2015
Version of record online: 11 August 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Siobhan O. Burns. 2016. Dendritic cell defects in primary immunodeficiency disorders. LymphoSign Journal.
3(1): 1-12. https://doi.org/10.14785/lpsn-2015-0001
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
