Human congenital T-cell receptor disorders
Abstract
Immunodeficiencies of most T-cell receptor (TCR) components (TCRID) have been reported in almost 40 patients worldwide who have also, at times, shown signs of autoimmunity. We updated their clinical, immunological, and molecular features with an emphasis on practical diagnosis, as the range of the disorder grows in complexity with new partial defects. Cellular and animal models are also reviewed and in some cases reveal their limitations for predicting TCRID immunopathology.
Introduction
T-cell receptors (TCR)
Mature T lymphocytes detect antigens with a variable surface TCR heterodimer, which is either αβ or γδ (Figure 1).
Figure 1:
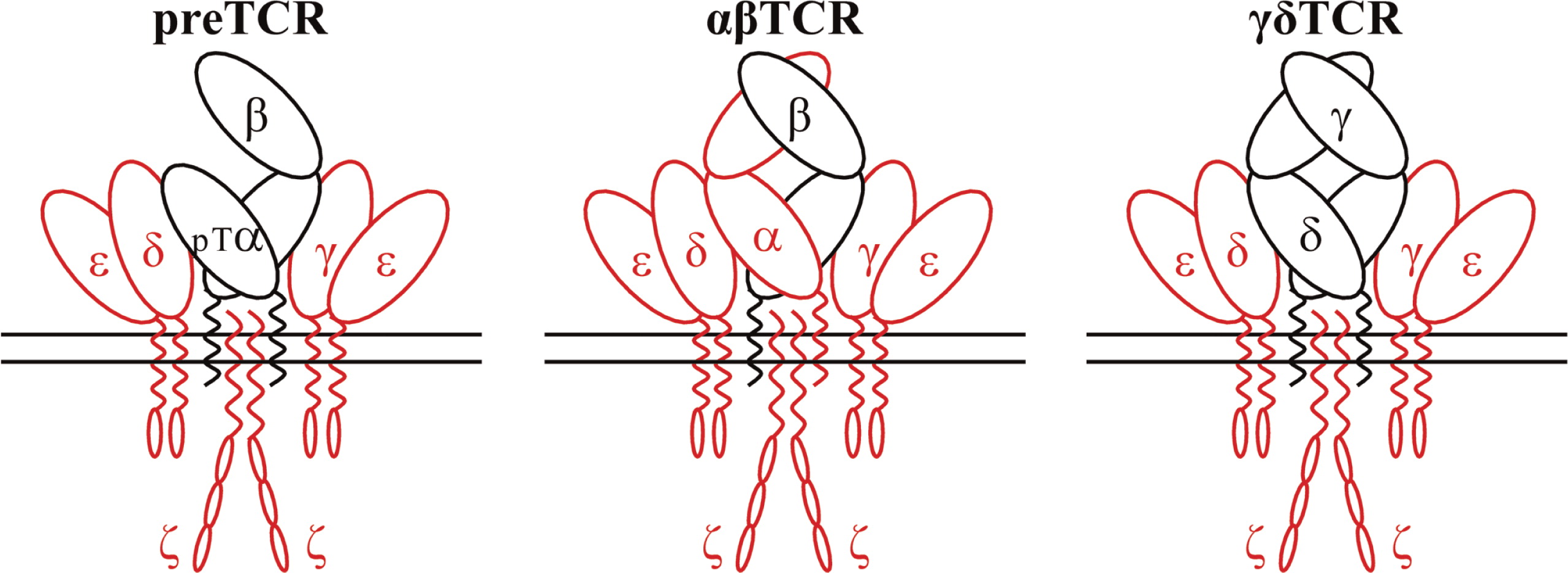
During early T-cell development, the invariant pre-Tα chain is used instead of TCRα. In humans, the variable TCR heterodimers form a complex with 2 invariant heterodimers termed CD3γɛ and CD3δɛ and a single invariant homodimer termed ζ or CD247 (Call et al. 2002). These 3 invariant dimers participate in assembly and surface expression of the whole TCR–CD3–CD247 complex, which we will refer to here as TCR. When a TCR interacts with its specific antigen through the variable heterodimers, intracellular signals are transduced to drive T-cell maturation or apoptosis in the thymus and T-cell activation, proliferation, and effector function or anergy–apoptosis in the periphery (Brownlie and Zamoyska 2013). The cytoplasmic tails of CD3ɛ and CD247 then expose their ITAM sequences (Love and Hayes 2010), which are phosphorylated by Lck, thus allowing the recruitment and Lck-mediated activation of ZAP-70. Activated ZAP-70 phosphorylates LAT, which recruits numerous signaling molecules to form a multiprotein complex termed the LAT signalosome. This signalosome, in turn, propagates signals to 3 major signaling pathways, namely MAPK, PI3K/AKT, and PLCγ-1, leading to the activation of transcription factors that are critical for gene expression and essential for T-cell growth and differentiation. Signals initiated from the TCR also result in actin reorganization and the activation of integrins by inside-out signaling (Kuhns and Davis 2012). Most αβTCR-bearing T cells recognize processed peptides associated with major histocompatibility complex (MHC) molecules, whereas γδTCR-bearing T cells can respond to a wide variety of antigens irrespective of their molecular nature including peptide and nonpeptide antigens, MHC-related proteins, phosphoantigens, and soluble proteins (Chien et al. 2014).
T-cell receptor immunodeficiencies (TCRID)
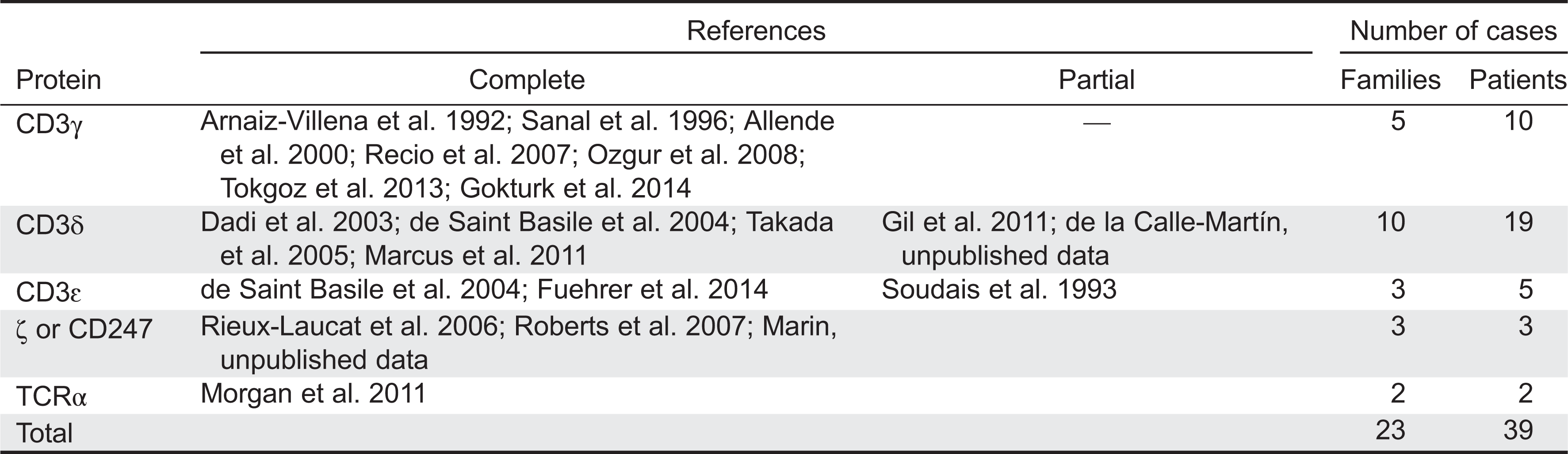
TCRIDs are low-prevalence autosomal recessive diseases characterized by impaired surface TCR expression, frequently associated with peripheral blood T lymphocytopenia, severe combined ID (SCID) and (or) autoimmune symptoms, but not associated with B or natural killer (NK) lymphocytopenia. The first TCRID was a human familial CD3 expression deficiency in a child with SCID and in his healthy sibling (Regueiro et al. 1986), later shown to be due to a complete CD3γ deficiency (Arnaiz-Villena et al. 1992). A second TCRID was soon reported (Thoenes et al. 1992) and shown to be due to partial CD3ɛ deficiency (Soudais et al. 1993). Several CD3, CD247, and TCR deficiencies have since been described (Figure 1, Table 1), which can be classified as complete or partial according to the absence or presence of residual levels of the affected protein. Although rare and sometimes based on single cases, TCRID offer rich information about the underpinnings of human TCR structure and function, which in turn impact our understanding of T-cell development and function. Therefore we, as have others (Casanova et al. 2014), believe they merit a detailed study.
Table 1:
Clinical and pathological manifestations
TCRID
Most patients show SCID or CID features such as recurrent respiratory infections, otitis, candidiasis, diarrhea, failure to thrive, and sometimes autoimmune phenomena in the first year of life. Thymus size can be small (CD3γ, δ, or ɛ deficiency (Arnaiz-Villena et al. 1991; de Saint Basile et al. 2004; Takada et al. 2005)) or normal (CD3γ or CD247 deficiency, unpublished). Chronic pyogenic infections, dysmorphic features, or bone abnormalities were not reported. Stem cell transplantation is indispensable when SCID is present, otherwise most patients die early in life as a consequence of viral infection (cytomegalovirus mainly, but also adenovirus). Autoimmune phenomena have been described in several patients, mainly when T cells are present. Most CD3γ-deficient patients showed autoimmune hemolytic anemia (AHA) or vitiligo (Arnaiz-Villena et al. 1992) and also autoimmune thyroiditis, thrombocytopenia, or autoimmune hepatitis (Gokturk et al. 2014). Two TCRα-deficient patients presented with vitiligo, AHA, or autoantibodies (Morgan et al. 2011). These results indicate that both positive selection and negative selection, which are TCR-dependent events, can be impaired in TCRID.
The CD3γ deficiency exception
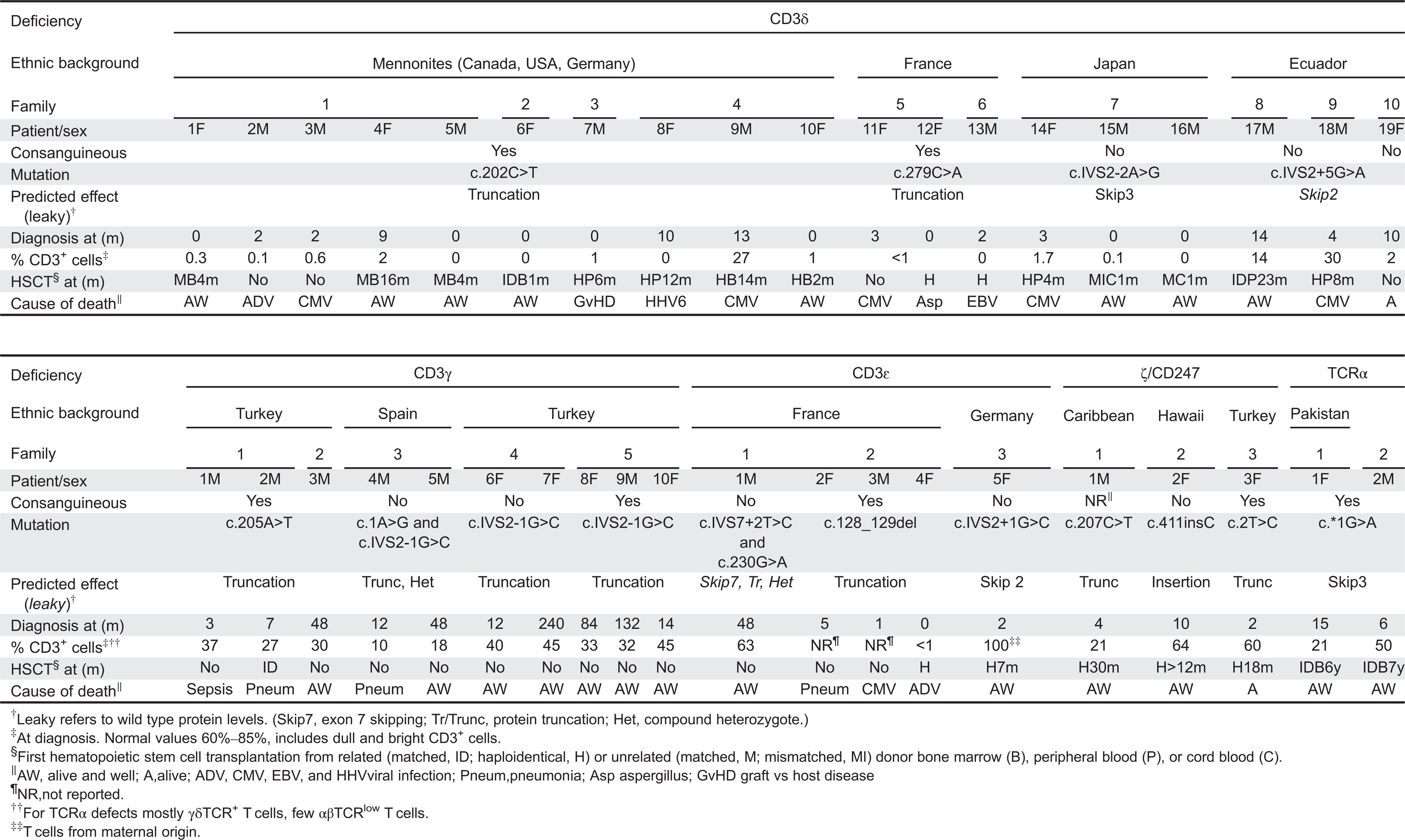
In contrast with all other reported TCRID due to complete protein deficiencies (Table 1), most CD3γ deficient individuals reported to date (7 out of 10) are presently alive and well without transplantation (Table 2), including some in their thirties. Late diagnosis, mild T lymphopenia, low TCR expression, and autoimmune features are common (see “TCRID” section). These limited immunological consequences are at odds with what is observed when the very homologous CD3δ chain is absent: early diagnosis, severe T lymphopenia, no TCR expression, and urgent transplantation. This clearly shows that CD3γ is not critical for survival in most cases, as compared with other TCR proteins, and particularly with CD3δ. The most likely molecular explanation is that CD3δ can replace CD3γ in the TCR, as has been reported (Zapata et al. 2004), whereas the reverse is not possible. As we review in the “Animal models” section, this is not the case in CD3γ Knock-Out (KO) mice. However, 3 out of 10 CD3γ-deficient patients, even though they had received transplants, did not survive to adulthood, thus suggesting that therapeutic decisions must be taken on a case by case basis.
Table 2:
Diagnosis
Differential diagnosis
A male or female infant showing T lymphopenia could be a result of TCRID, but also of mutations in IL7Rα, FOXN1, Coronin-1A, Zap70, TAP, MHC class II, transcription factors, PNP, ADA, or 22q11.2 (DiGeorge syndrome) (Al-Herz et al. 2014); however, only TCRID show TCR expression defects when T cells are present. Thus, a careful flow cytometry study is essential for the early differential diagnosis of TCRID, which is critical for patient survival, and should include both αβ and γδ T-cell subsets.
Sample quality
Samples are often received from abroad and travel long distances at changing temperatures before they reach the diagnostic lab. In such cases it is important to include age-matched controls that suffer similar shortcomings, so that comparisons can be meaningful.
T-cell subset definition and lymphopenia
TCRID may have no T cells and are thus properly called T−B+NK+. But often some T cells are present (T±B+NK+), although their identification using CD3 as an extracellular marker can be difficult owing to their reduced TCR expression. Indeed, some authors have reported T−B+NK+ phenotypes despite the presence of abundant CD4+ and CD8+ lymphocytes expressing very low levels of surface CD3 (Roberts et al. 2007). As intracellular CD3 is not normally used for screening TCRID, we used extracellular CD4 and CD8 to identify T cells in TCRID patients. We have shown that essentially all CD4+ and CD8bright cells within the lymphocyte gate are αβ T cells and, conversely, most double negative cells (DN, CD4−CD8−) in the same gate are γδ T cells (Muñoz-Ruiz et al. 2013). CD8dull lymphocytes are mostly NK cells. For comparative purposes, we have considered CD3+ lymphocytes in Table 2 because they are widely reported, although it is likely an underestimation of the true number of T cells. Chimerism should be excluded using standard fingerprinting procedures, to avoid for instance counting the mother's T cells, as reported recently in a new case of CD3ɛ deficiency (Fuehrer et al. 2014).
Using these criteria, most patients with complete defects show selective peripheral blood T lymphopenia, either T−B+NK+ or T±B+NK+ depending on the affected TCR chain, which is a first clue to the molecular basis of the pathology. T−B+NK+ has been associated with complete CD3ɛ or CD3δ defects, with less than 2% peripheral blood T cells. T±B+NK+ has been reported in complete CD3γ, TCRα, or CD247 defects. All partial TCRID show a T±B+NK+ or even a T+B+NK+ immunophenotype.
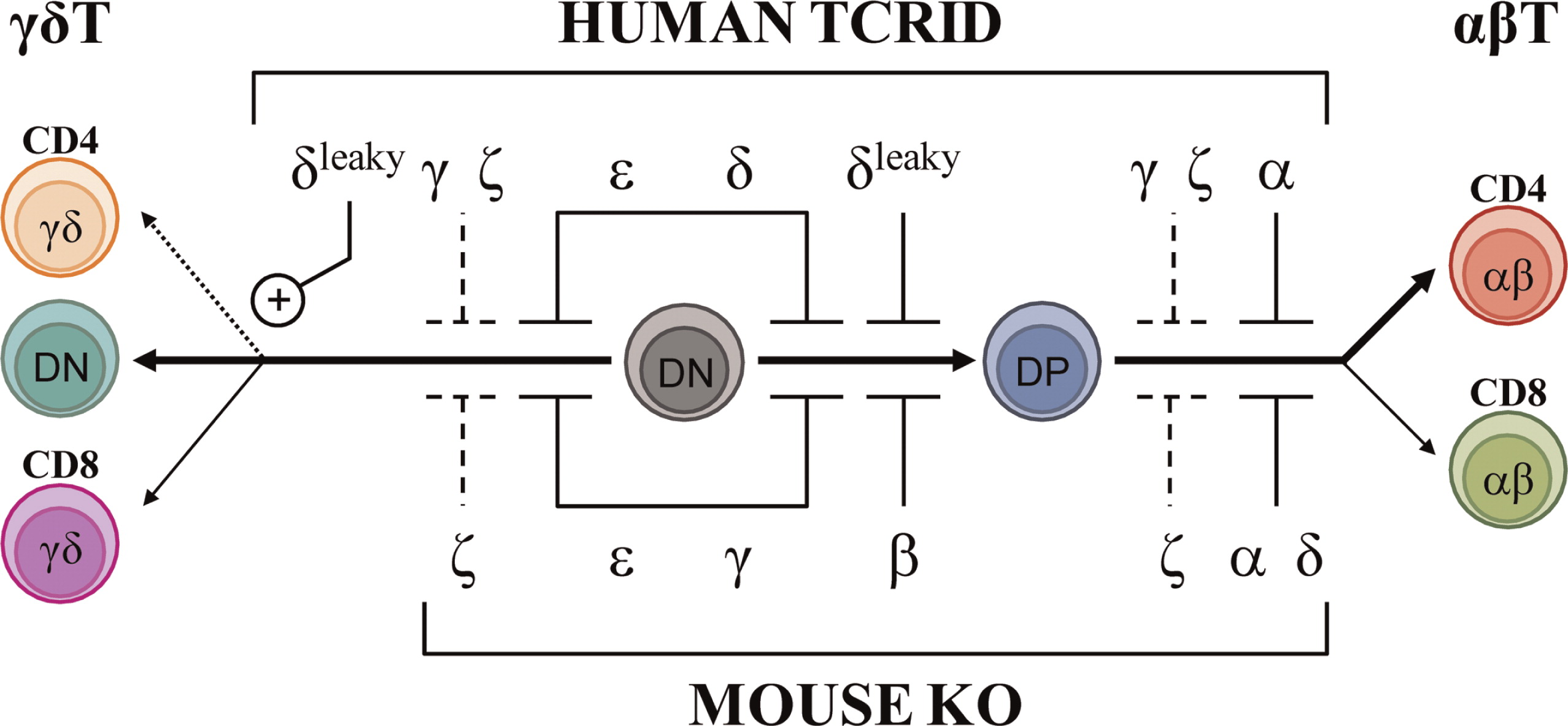
TCRID are best diagnosed if both αβ and γδ T cells are studied, as shown for complete TCRα deficiency (Morgan et al. 2011) and for partial CD3δ deficiency (Gil et al. 2011), both of which shared a Tαβ−γδ+B+NK+ immunophenotype despite their disparate molecular basis. Figure 2 (top) summarizes the effects of TCRID mutations in T-cell development.
Figure 2:
Surface TCR expression in patients’ lymphocytes
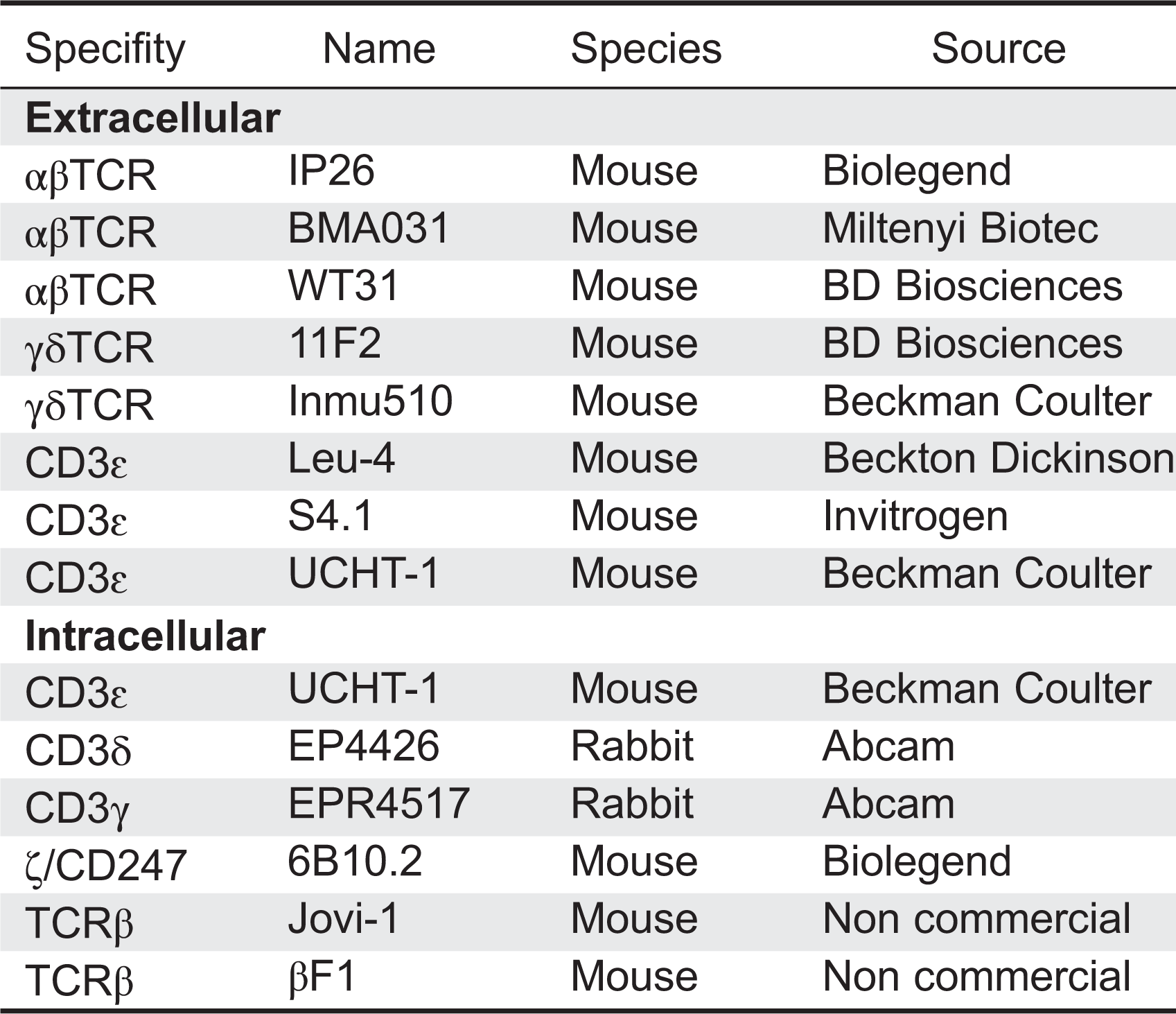
We have listed the monoclonal antibodies used in our lab for surface staining of peripheral blood lymphocytes from possible TCRID patients (Table 3, extracellular).
Table 3:
We start with just UCHT-1 (anti-CD3ɛ), 11F2 (anti-γδTCR), and any anti-CD4 and anti-CD8α antibodies, and in a different staining IP26 (anti-αβTCR) and 11F2 (anti-γδTCR). Within a subset (for instance, CD4+ lymphocytes), TCR Mean Fluorescence Intensity is evaluated with any of those CD3- or TCR-specific antibodies in comparison with that of an age-matched healthy control (Figure 3, top center). The expression of CD4 itself can serve as an internal control (Figure 3, top left). We normally use a range of CD3-specific monoclonal antibodies, such as those listed in Table 3 (extracellular), to confirm any TCR expression defect.
Figure 3:
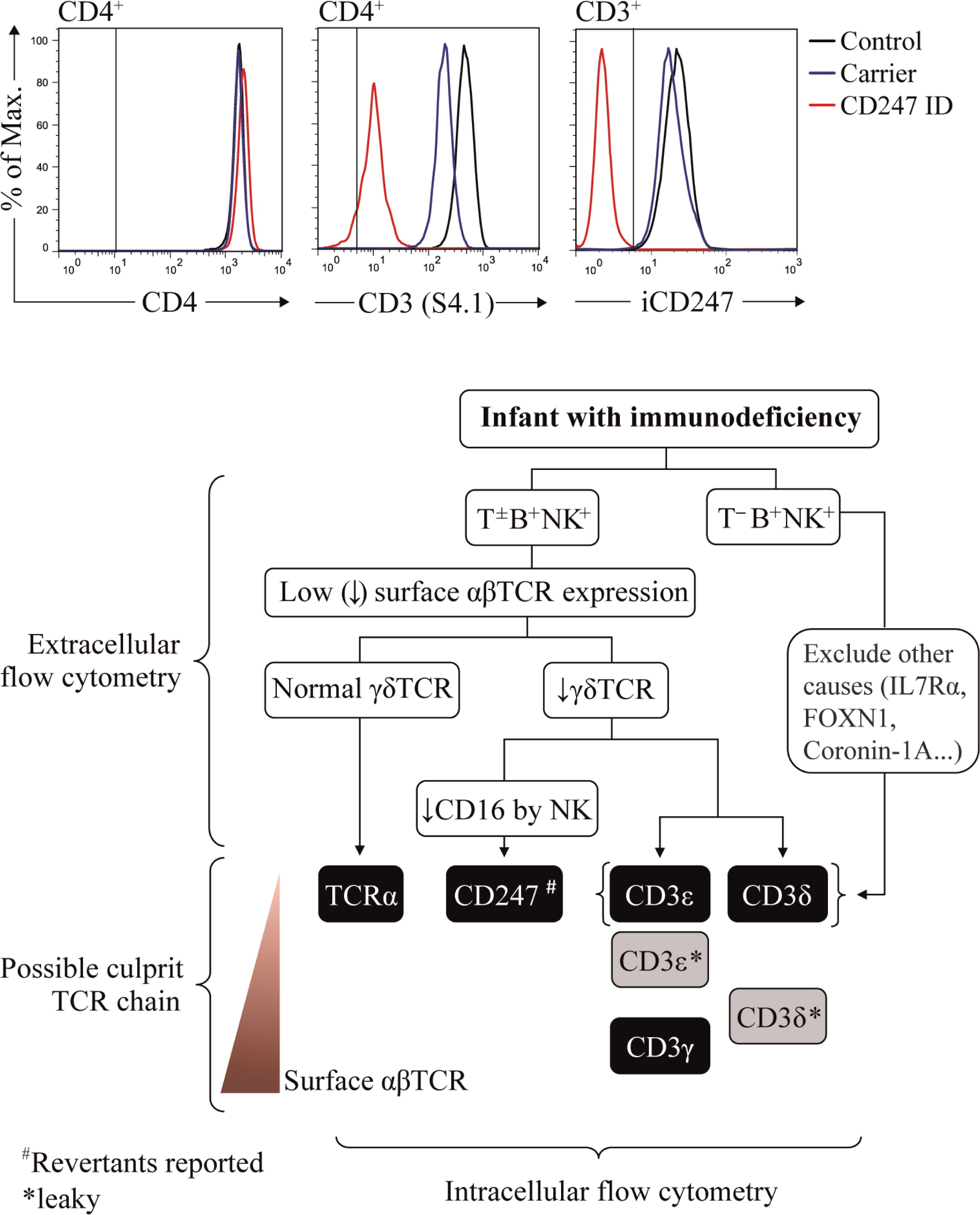
Family members are not appropriate controls, because in some cases they also show (partial) TCR expression defects that may underestimate the patient's defect (see Figure 3 and Muñoz-Ruiz et al. 2013, top center and “Surface TCR expression in carriers’ lymphocytes”). If no TCR expression defect is observed, TCRID can be excluded. If a TCR expression defect is ascertained, both αβ and γδ T cells should be analyzed. If αβ T cells (defined as CD4+ and (or) CD8bright) show impaired TCR expression and γδTCR+ show normal surface TCR expression, TCRα deficiency is likely. If αβ and γδ T cells share a surface TCR expression defect, a CD3 or CD247 defect may be suspected and further studies are required to identify the culprit protein (see “Protein identification in patients’ lymphocytes”), as indicated in the diagnostic flow chart (Figure 3, bottom). The following TCR expression hierarchy may be useful for the diagnosis of TCRID due to the lack of the indicated chains: CD3ɛ=CD3δ=TCRα≥CD247>CD3γ (our experience). Also, studying NK cells can be informative, as CD247 deficiency affects the expression of NK surface receptors such as CD16 or NKp30 (Reyburn, unpublished data). It is important to note that in contrast to αβ T cells, the range of surface TCR levels in γδ T cells in different individuals is quite heterogeneous (Garcillán et al. 2014), a fact that must be taken into account when analyzing the overlays of γδ T cells.
Lastly, it is advisable to grow T cells using IL-2 plus allogeneic feeder cells or immortalize them with Herpesvirus saimiri (HVS) or Human T-cell leukemia virus type 1 (HTLV-1), (see “Functional studies” and “Cellular models”), so that the detected TCR expression defects can be ascertained and further analyzed.
Surface TCR expression in carriers’ lymphocytes
The detection of TCRID mutation carriers using flow cytometry is difficult because surface TCR expression is often within the normal range. Again, overlays of extracellular CD3ɛ staining can be used to compare several family members, as shown in Figure 3 (top center), for CD247 (Marin and Garcillán, unpublished data). Using similar strategies, we have shown that CD3γ+/−, CD3δ+/− (and even CD3δ+/leaky) carriers can be identified, although the difference is not so clear and can be confounded by normal surface TCR variation (Muñoz-Ruiz et al. 2013). Of course, a genetic analysis is required for confirmation.
Protein identification in patients’ lymphocytes
In the past, immunoprecipitation (Perez-Aciego et al. 1991) or Western blot techniques (Gil et al. 2011) had to be used for the identification of protein defects. But these approaches require large amounts of T cells, which are difficult to obtain when samples are scarce (as is often the case with infant patients), and even more difficult in cases with severe T-cell lymphopenia. Generation of T-cell lines can be a solution, but they take precious time, thereby delaying diagnosis. CD3 chain-specific antibodies for intracellular staining have become available lately, and in our hands simplified the diagnosis of TCRID, which can now be done rapidly. Table 3 (intracellular) lists such antibodies and Figure 3 (top right) shows a recent example of their use. Once the culprit protein is defined, genetic validation is indispensable to establish the molecular basis of the protein defect.
In summary, excellent reagents are now available to diagnose TCRID defects in a record time, even when very few T cells are present.
Mutation analysis
A mutation database has been established that contains most described mutations in the genes encoding for TCR chains (Piirilä et al. 2006). Mutations causing TCRID include missense, nonsense, and splice-site mutations as well as less common genomic deletions and insertions (Table 4). Figure 4 illustrates their presumed protein products that, when truncated, are often unstable.
Figure 4:
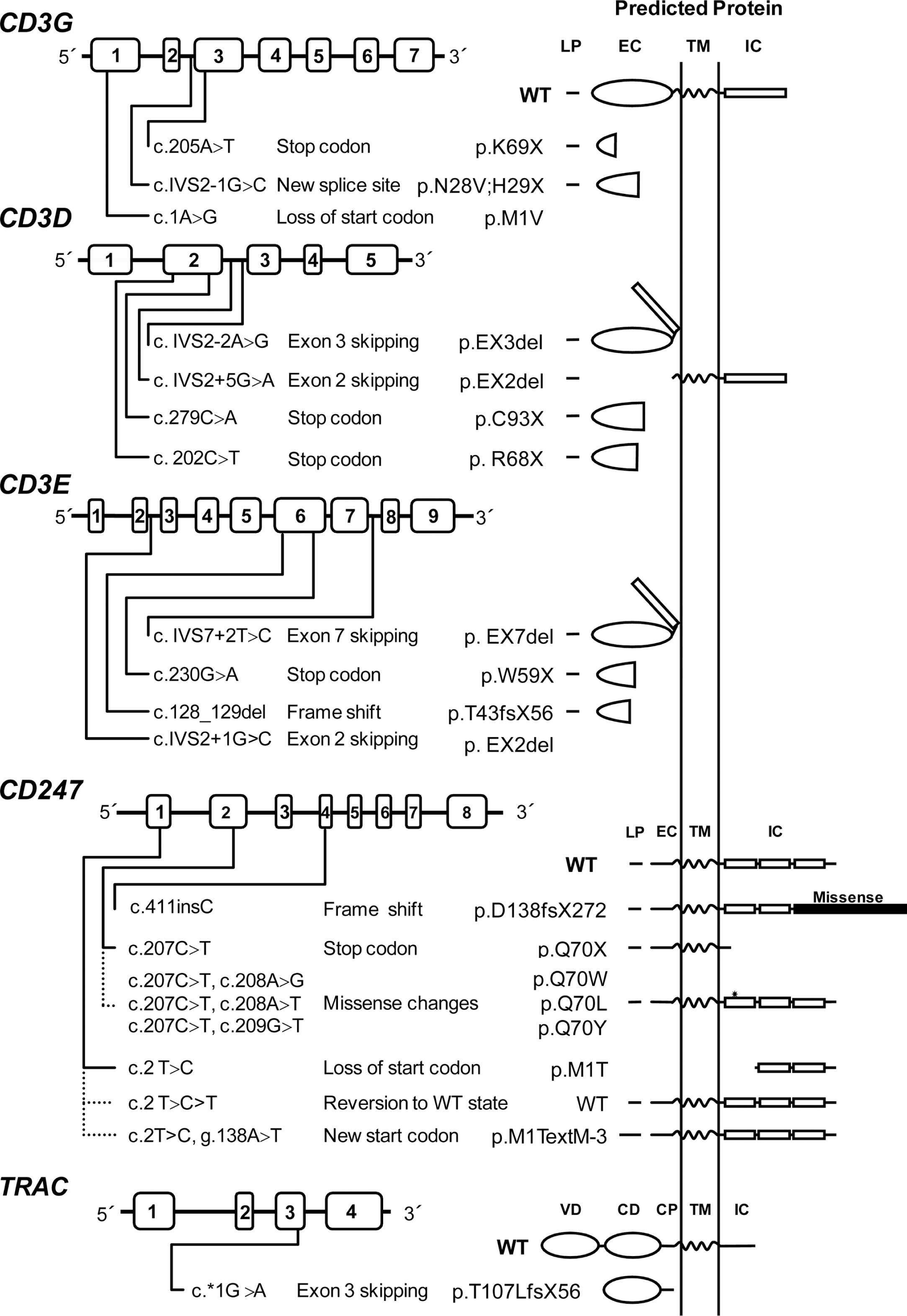
Table 4:
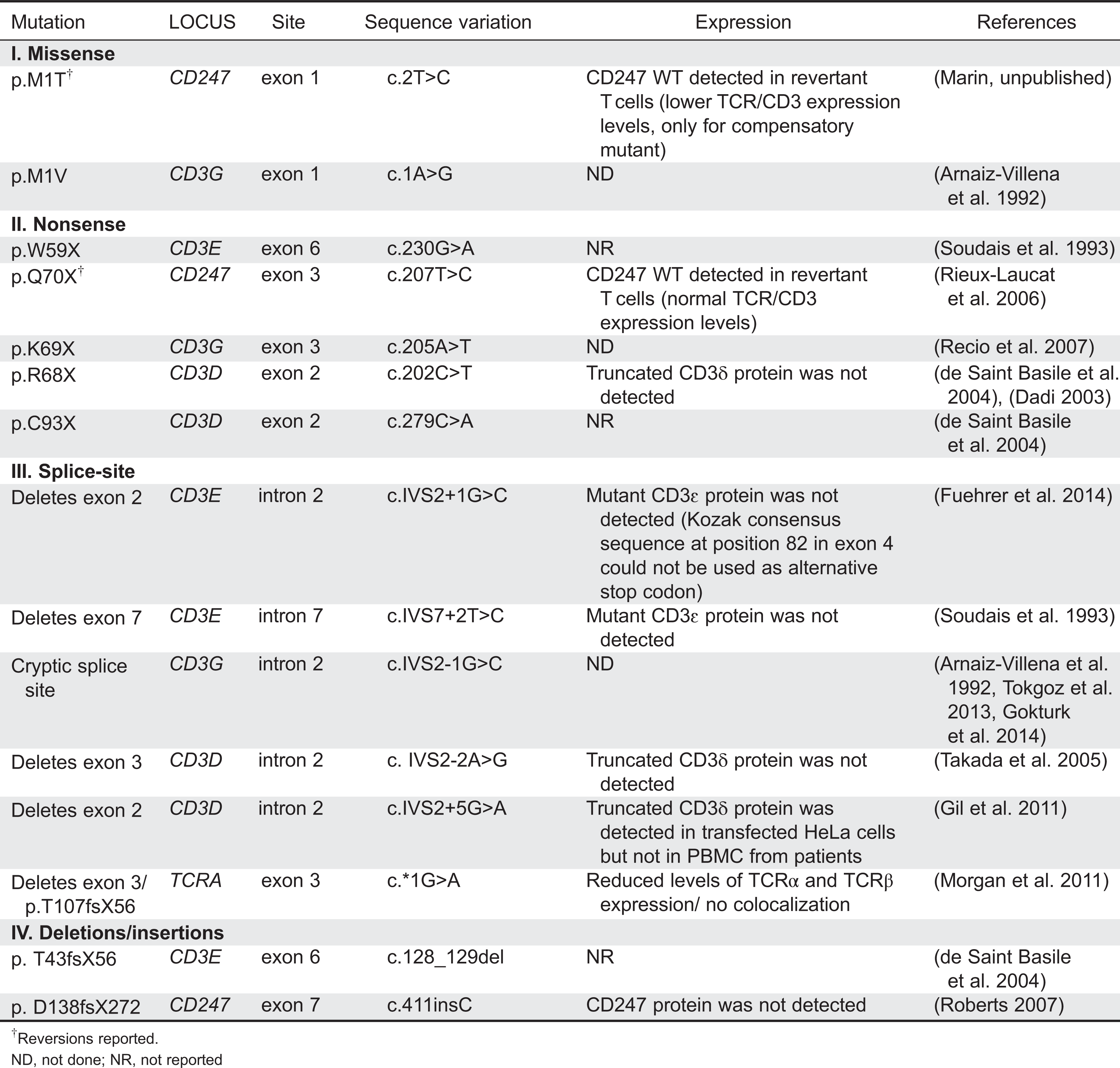
Most mutations drastically affect the expression of the protein (complete defects), whereas in some leaky splice-site mutations, small amounts of normal transcripts were produced, allowing for the expression of some normal protein and thus causing a partial phenotype. This is the case for CD3δ and CD3ɛ TCRID.
Potentially truncated proteins have also been reported and were likely unstable, as they were undetectable in T cells. Indeed, the in-frame deletion of exon 7 predicted a mutant CD3ɛ protein that was not found even with very sensitive techniques (Thoenes et al. 1992). Also, a predicted mutant CD3δ protein selectively lacking the extracellular domain encoded by exon 2 was undetectable in peripheral blood mononuclear cells (PBMC) from the patient or carriers, although it could be shown in transfected non-T cells (Gil et al. 2011). This may be mutation specific, as shown recently in a new splicing mutation in CD3E intron 2, which was predicted to delete exon 2 and thus the start codon. As a consequence, no protein was detected by Western blot either in carrier PBMC or in transfected non-T cells (Fuehrer et al. 2014). In TCRα deficiency, a c.*1G>A mutation resulted in an aberrant transcript joining exon 2 to the normally untranslated exon 4. In the predicted translation product, the 35 C-terminal amino acids would be replaced by 56 amino acids encoded by exon 4. This would result in partial loss of the connecting peptide domain and abolition of the transmembrane and cytoplasmic domains of the TCRα chain. In PBMC, no TCRα or TCRβ proteins were detected by Western blotting with specific antibodies, but minute amounts of both chains were detectable by immunofluorescence microscopy using a different set of antibodies. The authors concluded that the residual predicted truncated TCRα failed to bind normally to TCRβ, because in the latter experiment the TCR chains did not colocalize. This was later confirmed in transfected HeLa cells (Morgan et al. 2011).
Nonsense mutations are clearly deleterious and frequently associate with severe TCRID immunophenotypes, except for complete CD3γ and partial CD3ɛ deficiencies. In CD3δ deficiency, a microarray analysis in thymocytes revealed more than 2-fold lower specific transcripts, but no CD3δ protein was detected by Western blot analysis of the same lysates or by thymus immunohistochemistry (Dadi et al. 2003).
Somatic mosaicism caused by reversions can ease otherwise severe clinical phenotypes. Reversions have been reported in several disorders, including dermatological, metabolic, and immunological disorders such as ADA deficiency, X-linked SCID, and Wiskott–Aldrich syndrome (Hirschhorn 2003; Wada and Candotti 2008).
Reversion of some T cells to carrier TCR expression levels has been shown to be the consequence of CD247 germ-line mutations when studied (Figure 4). However, TCR signaling after stimulation with phytohemagglutinin (PHA) or anti-CD3 of revertant T cells can be impaired (Rieux-Laucat et al. 2006) or restored to carrier levels (Marin, unpublished data) for reasons yet to be defined. Interestingly, no reversions have been reported in other TCRID, suggesting that CD247 is more prone to somatic mutations than the CD3 or TRAC genes.
CD3GDE haplotype analysis using polymorphic markers can be useful (i) to rule out CD3 mutations, if segregation does not fit the pedigree; (ii) to establish the existence of founder alleles even when families are unaware of such connections, as we have reported for CD3G in Turkey (Recio et al. 2007) and for CD3D in Ecuador (Gil et al. 2011); and (iii) for carrier detection and (or) prenatal diagnosis, because recombination within the CD3 gene complex is rare.
Screening tests specific for some founder CD3G and CD3D mutations using TaqI and BsaAI (Table 5) have been developed for several Turkish and Ecuadorian families, respectively, which help to reach quick diagnoses and segregation data for genetic counselling (Recio et al. 2007; Gil et al. 2011). Therefore, in Turkish or Ecuadorian patients with an immunophenotype suggestive of CD3γ or CD3δ deficiency, regardless of the accompanying spectrum of clinical symptoms, screening for such mutations should be considered in their molecular diagnosis.
Table 5:

Functional studies
T-cell functional analyses both in vivo and in vitro are useful, but not critical, for TCRID diagnosis, as they are frequently impaired even when T cells are abundant or in healthy siblings, as reported for CD3γ deficiencies (Recio et al. 2007). More often, lymphopenia precludes functional studies in primary T cells, so it becomes necessary to use immortalized cell lines to explore the functional impact of the mutations in T-cell function. For this purpose, T cells can be immortalized with HVS or HTLV-1 (Pacheco-Castro et al. 1998). Additionally, fresh PBMC can be cultured with allogeneic feeder cells (a γ-irradiated mixture of EBV-transformed lymphoblastoid cells and PBMC from several healthy donors).
TCR-triggered early activation events (such as Ca2+ influx and CD69 or CD25 induction) and late activation events (such as proliferation induced by mitogenic, antigenic, or allogeneic stimuli) sometimes show contradictory results. For instance, T cells from CD3δleaky patients showed strongly reduced induction of CD69 and CD25 but normal proliferation when anti-CD3 or PHA stimulation were used (Gil et al. 2011). In other cases, although anti-CD3 responses were reduced, co-stimulation with CD28 or IL-2 restored proliferative responses (Le Deist et al. 1991; Rieux-Laucat et al. 2006).
Thymus function, often measured as output of peripheral blood recent thymic emigrants (CD4+CD45RA+CD31+ lymphocytes), is frequently impaired in TCRID. Additionally, TCR diversity is commonly poor, as measured looking at TCRB clonality and TCRVβ diversity (Gil et al. 2011).
Despite impaired T-cell responses, TCRID are not always associated with diminished B-cell memory subsets, reduced immunoglobulin (Ig) levels, or abnormal vaccination responses, as reported in TCRα deficiency (Morgan et al. 2011) and in CD3δleaky deficiency (Gil et al. 2011).
Treatment and prognosis
The only effective treatment is hematopoietic stem cell transplantation (HSCT), and the sooner the better (Marcus et al. 2011). Patients with successful transplants have been shown to be healthy up to 18 years post-transplantation. Otherwise, the prognosis is very poor for those with complete or even some partial defects such as CD3δleaky (Table 2), with some exceptions (see next paragraph). Matched related, haploidentical mismatched related, matched unrelated, and mismatched unrelated donors have all been used for HSCT, generally after myeloablative conditioning, but matched related donor HSCT is also used without conditioning when SCID is diagnosed. Complete T-cell reconstitution is generally observed (including normalized B cell function), but split-chimerism has been recently reported for CD3ɛ deficiency, which 15 years later resulted in the need of intravenous Igs owing to of lack of switched memory B cells (Fuehrer et al. 2014). A diminished switched memory B-cell subset is a common finding in other SCID patients (Sarantopoulos et al. 2009). A frequent complication of HSCT is graft versus host disease, which can be treated with immunosuppression as was done recently in a case with CD247 deficiency (Aydogmus, unpublished data). When autoimmunity is present, immunosuppression is also required (steroids and cyclosporin A), as reported recently for Evans syndrome in CD3γ deficiency (Tokgoz et al. 2013).
Some CD3γ- and a partial CD3ɛ-deficient patient without ID symptoms did not receive HSCT, but rather antibiotics only when symptoms developed (Allende et al. 2000) or prophylactic intravenous Igs with antibiotics (Le Deist et al. 1991), respectively.
Models of TCRID
Animal models
Knock-Out (KO) mouse models (Table 6) have been used to replicate human congenital TCRID, because mouse and human TCR and CD3 chains share significant sequence and functional homology (Dave 2009). Indeed, a block in intrathymic T-cell development and reduced TCR expression was reported in all cases, although sharp differences were found for CD3γ and CD3δ (Figure 2).
Table 6:
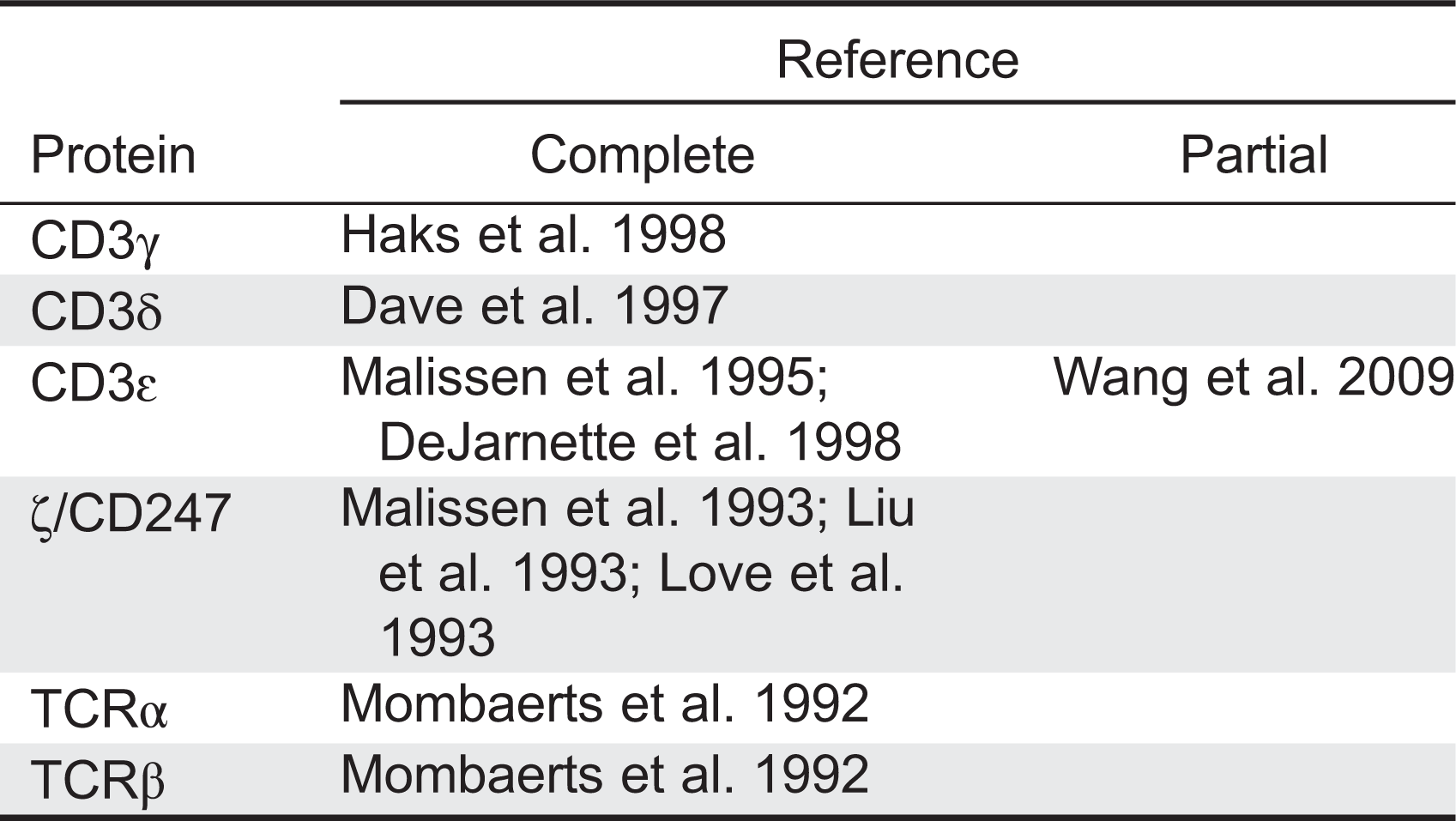
CD3δ−/−mice and humans shared a severe selective αβ T lymphocytopenia, but this was due to a block at different checkpoints (DP in mice vs DN in humans, Dave 2009). However, although CD3δ-deficient patients also lacked γδ T cells, CD3δ−/−mice showed no obvious defect in γδ T-cell development or TCR expression (Dave et al. 1997). The mechanism became clear when stoichiometry studies showed that in humans, but not in mice, the γδTCR incorporates CD3δ (Hayes and Love 2006; Siegers et al. 2007).
CD3γ−/−mice showed very few peripheral T cells (around 10% of normal values) with low surface TCR and abnormal stoichiometry (αβ/δɛδɛζζ) owing to a severe block at the DN stage (Haks et al. 1998). By contrast, CD3γ deficiency in humans allows the selection of substantial numbers of peripheral T cells (Recio et al. 2007), which in the case of αβ T lymphocytes expressed relatively high levels of functional αβTCR complexes (Pacheco-Castro et al. 1998), albeit with an abnormal stoichiometry (αβTCR/CD3δɛδɛζζ) and an impaired association to CD247 (Zapata et al. 2004). Therefore, the lack of CD3γ or CD3δ during αβ T-cell development gives rise to inverted phenotypes in mice versus humans, often precluding a meaningful comparison (Siegers et al. 2007).
CD3δ and CD3γ are the most homologous CD3 proteins. Yet, although patients lacking CD3δ are devoid of mature T cells, CD3γ-deficient individuals do have seemingly functional αβ and γδ T lymphocytes and generally less severe clinical features (Table 2). These differential phenotypes indicate that in humans CD3δ replaces CD3γ in the pre-TCR and TCR for stable expression on and signalling from the cell surface, allowing thymic development of fairly functional T cells. In support of this notion, expression of a human, but not a mouse, CD3δ transgene rescued αβ and γδ T-cell development in mice that were deficient for both CD3δ and CD3γ (Fernández-Malavé et al. 2006; Siegers et al. 2007). Because pre-TCR and γδTCR complexes in mice do not seem to incorporate CD3δ (Malissen et al. 1999), these data further suggest that CD3δ has evolved distinct features in humans that allows it to substitute CD3γ in both species.
CD3ɛ−/−mice showed abrogated T-cell development with a severe block at the DN stage (DeJarnette et al. 1998) (Figure 2), supporting a shared critical role on thymocyte development versus humans (de Saint Basile et al. 2004).
As observed in humans, CD247−/−mice (Liu et al. 1993) showed a partial block at the DP stage with weak transition to SP T cells and very low surface TCR in both thymocytes and peripheral T cells, which are nonfunctional (Liu et al. 1993).
T-cell development in TCRα−/−mice is blocked at the DP stage, indicating that TCR gene rearrangements are critical for αβ T-cell differentiation, whereas γδ T cells are unscathed (Mombaerts et al. 1992).
Cellular models
The complex structure of the TCR and the severe T-cell lymphopenia normally associated to TCRID (Mazariegos et al. 2013) has hampered the dissection of the precise role of each TCR chain for TCR assembly and T-cell selection and function. KO mice, TCRID patients, mutagenized T cells lacking certain chains, TCR-transfected non-T cells and, more recently, Knocked-Down (KD) T cells tell different yet complementary tales in each species, as we review here for cellular models.
Human T cells
Jurkat mutants
Several variants of the αβ T-cell leukemia line Jurkat, lacking TCRα, TCRβ, or CD3γ have been obtained over the years (Table 7). All lacked surface TCR, thereby supporting the idea that these proteins are required for TCR expression.
Table 7:
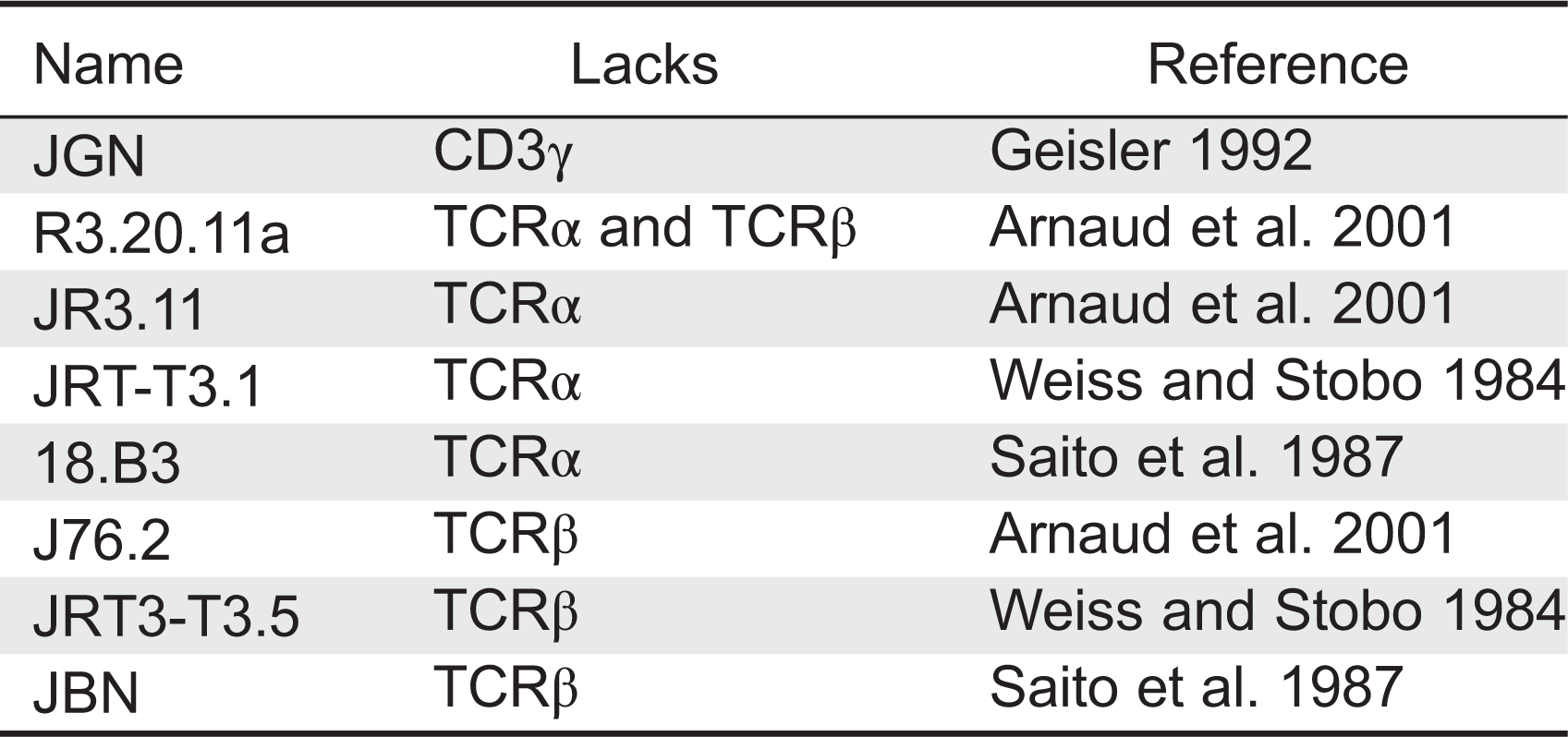
In 1992, Geisler described a Jurkat cell line, termed Jurkat Gamma Negative (JGN), deficient for surface TCR expression owing to the lack of CD3γ (Geisler 1992). Using JGN and site-directed mutagenesis, they identified a di-leucine motif in the intracellular (IC) domain of CD3γ implicated in TCR internalization (Dietrich et al. 1996a), two EC interaction sites of CD3γ with CD3ɛ, and one transmembrane (TM) residue involved in binding to TCRβ. They also demonstrated that N-glycosylation in CD3γ is not required for TCR assembly and expression, and that the IC domain of CD3γ is not required for surface TCR expression (Dietrich et al. 1996b). Experiments using chimeric constructions of CD3γ and CD3δ demonstrated that the IC and TM regions of these highly homologous proteins were interchangeable (Wegener et al. 1995). TCRα- or TCRβ-deficient Jurkat cell lines have been also used to define important assembly residues in TCR chains, TCR assembly hierarchy, endoplasmic reticulum retention of partial complexes, and degradation of TCR chains (Dietrich et al. 1999; Martin et al. 1999; Arnaud et al. 2001).
Knocked-Down (KD) Jurkat
We have generated Jurkat KD lines for CD3δ or CD3γ that show that both chains, although highly homologous, are essential for surface TCR expression (Garcillán, unpublished data). This observation is in agreement with the profound T-specific lymphopenia found in CD3δ KO mice and CD3δ-deficient patients, although residual surface TCR was found in T cells from both mutants. However, cellular models do not always recapitulate the human TCRID, as CD3γ-deficient patients (but not KO mice) showed abundant T cells and surface TCR expression (Perez-Aciego et al. 1991), whereas JGN lacked surface TCR. We believe that intrathymic selection pressure can explain this discrepancy, which also shows that human and murine TCR chains have acquired different roles upon speciation (see “Animal models”).
Immortalized T cells from TCRID patients
Both HVS- and HTLV-1-immortalized T-cell lines have been derived from CD3γ-deficient patients (Pacheco-Castro et al. 1998; Torres et al. 2003), which preserved the TCR expression features of their donors. These cell lines have been instrumental to show that despite decreased surface TCR expression several mature T-cell responses were normally induced via their mutant TCR, including calcium flux, cytotoxicity, up-regulation of CD69, proliferation, and synthesis of specific cytokines, such as TNFα and IFNγ. In contrast, TCR-induced IL-2 synthesis, adhesion, tyrosine phosphorylation, and PMA-induced TCR downregulation were impaired (Torres et al. 2002; Torres et al. 2003; Reine et al. 2011).
Cell lines derived from patients with partial CD3δ or complete CD247 deficiency have been also generated using allogeneic feeder cells and IL-2 (Gil et al. 2011; Garcillán et al. 2014; Marin, unpublished data). These cell lines also preserved the features of the primary lymphocytes and have been useful for biochemical and functional analyses.
Mouse T cells
Several murine T-cell lines deficient in one of the TCR chains have been also studied. In 1988, a CD247 deficient cell line (MA5.8) was generated by chemically mutagenesis of the antigen-specific 2B4 T-cell hybridomas (Sussman et al. 1988). MA5.8 cells were able to express small amounts of surface TCR but failed to respond normally to antigens. MA5.8 has been widely used to establish the role of CD247 in TCR assembly (Dietrich et al. 1999; Delgado and Alarcon 2005) and to study CD247 splicing variants present in systemic lupus erythematosus patients (Tsuzaka et al. 2003; Tsuzaka et al. 2005). Later, a CD3δ-deficient T-cell line was obtained by repetitive subcloning at limiting dilution of the 2B4 T-cell hybridoma line. Studies with this cell line showed that, in the absence of CD3δ, surface TCR expression was abolished and the other TCR chains were retained in the endoplasmic reticulum (Bonifacino et al. 1989). Another study corroborated these results analyzing a murine cytotoxic T lymphocyte (CTL) clone that selectively lacked expression of the CD3δ mRNA (Buferne et al. 1992). Lastly, studies in a CTL clone deficient in both CD3δ and CD3γ showed that expression of either CD3δ or CD3γ alone failed to reconstitute surface TCR expression, but reconstitution with cytoplasmically truncated CD3δ and CD3γ led to reexpression of the TCR, demonstrating that both chains are required for surface TCR expression, whereas their cytoplasmic domains are dispensable (Luton et al. 1997).
A controversy about the requirement of CD3γ or CD3δ for TCR assembly and surface expression has been active for years. Several studies using primary cells, cell lines or TCR-transfected non-T cells have led to different conclusions, both for and against their critical role (Table 8).
Table 8:

ACKNOWLEDGEMENTS
We thank all patients for their cooperation and Drs C. Aydogmus and F. Cipe (Pediatric Immunology, Istanbul Kanuni Sultan Süleyman Hospital, Istambul, Turkey), A. Ikinciogullari, S. Haskologlu and F. Dogu (Pediatric Immunology-Allergy, Ankara University School of Medicine, Ankara, Turkey), G. Esteso, M. Valés-Gómez and H.T. Reyburn (Immunology and Oncology, National Centre for Biotechnology CSIC, Madrid, Spain), J. Gil (Immunology, Hospital Gregorio Marañón, Madrid, Spain), O. de la Calle (Immunology, Hospital San Pau, Barcelona, Spain), R. Cedeño (Hospital Oncológico “Dr Julio Villacreses Colmont” Solca Manabi-Esmeraldas, Portoviejo, Manabi, Ecuador), and M. Hönig (Immunologie, Universitätsklinik für Kinder- und Jugendmedizin, Ulm, Germany) for sharing unpublished data.
This work was supported by grants from MINECO (SAF2011-24235 and BES-2012-055054), CAM (S2010/BMD-2316), RIER (RD08-0075-0002) and Lair (2012/0070).
REFERENCES
Al-Herz W., Bousfiha A., Casanova J.L., Chatila T., Conley M.E., Cunningham-Rundles C., Etzioni A., Franco J.L., Gaspar H.B., Holland S.M., Klein C., Nonoyama S., Ochs H.D., Oksenhendler E., Picard C., Puck J.M., Sullivan K., and Tang M.L. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency Front Immunol. 2014 5 162
Allende L.M., Garcia-Perez M.A., Moreno A., Ruiz-Contreras J., and Arnaiz-Villena A. Fourteen years’ follow-up of an autoimmune patient lacking the CD3 gamma subunit of the T-lymphocyte receptor Blood. 2000 96 12 4007 -4008
Arnaiz-Villena A., Perez-Aciego P., Ballestin C., Sotelo T., Perez-Seoane C., Martin-Villa J.M., and Regueiro J.R. Biochemical basis of a novel T lymphocyte receptor immunodeficiency by immunohistochemistry. A possible CD3 gamma abnormality Lab Invest. 1991 64 5 675 -681
Arnaiz-Villena A., Timon M., Corell A., Perez-Aciego P., Martin-Villa J.M., and Regueiro J.R. Brief report: primary immunodeficiency caused by mutations in the gene encoding the CD3-gamma subunit of the T-lymphocyte receptor N. Engl. J. Med. 1992 327 8 529 -533
Arnaud J., Erard M., Martin E., Llobera R., Gouaillard C., Constans J., and Rubin B. Molecular modelling and endoplasmic reticulum retention of mutated TCR/CD3 complexes Scand. J. Immunol. 2001 53 6 540 -552
Bonifacino J.S., Suzuki C.K., Lippincott-Schwartz J., Weissman A.M., and Klausner R.D. Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly J. Cell Biol. 1989 109 1 73 -83
Brownlie R.J. and Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded Nat. Rev. Immunol. 2013 13 4 257 -269
Buferne M., Luton F., Letourneur F., Hoeveler A., Couez D., Barad M., Malissen B., Schmitt-Verhulst A.M., and Boyer C. Role of CD3 delta in surface expression of the TCR/CD3 complex and in activation for killing analyzed with a CD3 delta-negative cytotoxic T lymphocyte variant J. Immunol. 1992 148 3 657 -664
Call M.E., Pyrdol J., Wiedmann M., and Wucherpfennig K.W. The organizing principle in the formation of the T Cell Receptor-CD3 complex Cell. 2002 111 967 -979
Casanova J.L., Conley M.E., Seligman S.J., Abel L., and Notarangelo L.D. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies J. Exp. Med. 2014
Chien Y.H., Meyer C., and Bonneville M. gammadelta T Cells: first line of defense and beyond Ann. Rev. Immunol. 2014
Dadi H., Simon A.J., and Roifman C.M. Effect of CD3d deficiency on maturation of ab and gd T-cell lineages in Severe Combined Immunodeficiency N. Eng. J. Med. 2003 349 19 1821 -1828
Dave V.P. Hierarchical role of CD3 chains in thymocyte development Immunol. Rev. 2009 232 1 22 -33
Dave V.P., Cao Z., Browne C., Alarcon B., Fernandez-Miguel G., Lafaille J., de la Hera A., Tonegawa S., and Kappes D.J. CD3 delta deficiency arrests development of the alpha beta but not the gamma delta T cell lineage EMBO J. 1997 16 6 1360 -1370
de Saint Basile G., Geissmann F., Flori E., Uring-Lambert B., Soudais C., Cavazzana-Calvo M., Durandy A., Jabado N., Fischer A., and Le Deist F. Severe combined immunodeficiency caused by deficiency in either the delta or the epsilon subunit of CD3 J. Clin. Invest. 2004 114 10 1512 -1517
DeJarnette J.B., Sommers C.L., Huang K., Woodside K.J., Emmons R., Katz K., Shores E.W., and Love P.E. Specific requirement for CD3epsilon in T cell development Proc. Natl. Acad. Sci. USA. 1998 95 25 14909 -14914
Delgado P. and Alarcon B. An orderly inactivation of intracellular retention signals controls surface expression of the T cell antigen receptor J. Exp. Med. 2005 201 4 555 -566
Dietrich J., Hou X., Wegener A.M., Pedersen L.O., Odum N., and Geisler C. Molecular characterization of the di-leucine-based internalization motif of the T cell receptor J. Biol. Chem. 1996a 271 19 11441 -11448
Dietrich J., Kastrup J., Lauritsen J.P., Menne C., von Bulow F., and Geisler C. TCRzeta is transported to and retained in the Golgi apparatus independently of other TCR chains: implications for TCR assembly Eur. J. Immunol. 1999 29 5 1719 -1728
Dietrich J., Neisig A., Hou X., Wegener A.M., Gajhede M., and Geisler C. Role of CD3 gamma in T cell receptor assembly J. Cell Biol. 1996b 132 3 299 -310
Fernández-Malavé E., Wang N., Pulgar M., Schamel W.W., Alarcon B., and Terhorst C. Overlapping functions of human CD3delta and mouse CD3gamma in alphabeta T-cell development revealed in a humanized CD3gamma-mouse Blood. 2006 108 10 3420 -3427
Fuehrer M., Pannicke U., Schuetz C., Jacobsen E.M., Schulz A., Friedrich W., Schwarz K., and Honig M. Successful haploidentical hematopoietic stem cell transplantation in a patient with SCID due to CD3epsilon deficiency: need for IgG-substitution 6 years later Klin. Padiatr. 2014 226 3 149 -153
Garcillán B., Mazariegos M.S., Fisch P., Res P.C., Muñoz-Ruiz M., Gil J., Lopez-Granados E., Fernandez-Malave E., and Regueiro J.R. Enrichment of the rare CD4+γδ T-cell subset in patients with atypical CD3δ deficiency J. Allergy Clin. Immunol. 2014 133 4 1205 -1208.e9
Geisler C. Failure to synthesize the CD3-gamma chain. Consequences for T cell antigen receptor assembly, processing, and expression J. Immunol. 1992 148 8 2437 -2445
Gil J., Busto E.M., Garcillán B., Chean C., Garcia-Rodriguez M.C., Diaz-Alderete A., Navarro J., Reine J., Mencia A., Gurbindo D., Belendez C., Gordillo I., Duchniewicz M., Hohne K., Garcia-Sanchez F., Fernandez-Cruz E., Lopez-Granados E., Schamel W.W., Moreno-Pelayo M.A., Recio M.J., and Regueiro J.R. A leaky mutation in CD3D differentially affects alphabeta and gammadelta T cells and leads to a Talphabeta-Tgammadelta+B+NK+ human SCID J. Clin. Invest. 2011 121 10 3872 -3876
Gokturk B., Keles S., Kirac M., Artac H., Tokgoz H., Guner S.N., Caliskan U., Caliskaner Z., van der Burg M., van Dongen J., Morgan N.V., and Reisli I. CD3G gene defects in familial autoimmune thyroiditis Scand. J. Immunol. 2014
Haks M.C., Krimpenfort P., Borst J., and Kruisbeek A.M. The CD3gamma chain is essential for development of both the TCRalphabeta and TCRgammadelta lineages Embo. J. 1998 17 7 1871 -1882
Hayes S.M. and Love P.E. Stoichiometry of the murine gammadelta T cell receptor J. Exp. Med. 2006 203 1 47 -52
Hirschhorn R. In vivo reversion to normal of inherited mutations in humans J. Med. Genet. 2003 40 10 721 -728
Kappes D.J. and Tonegawa S. Surface expression of alternative forms of the TCR/CD3 complex Proc. Natl. Acad. Sci. USA. 1991 88 23 10619 -10623
Kuhns M.S. and Davis M.M. TCR signaling emerges from the sum of many parts Front Immunol. 2012 3 159
Le Deist F., Thoenes G., Corado J., Lisowska-Grospierre B., and Fischer A. Immunodeficiency with low expression of the T cell receptor/CD3 complex. Effect on T lymphocyte activation Eur. J. Immunol. 1991 21 7 1641 -1647
Liu C.P., Ueda R., She J., Sancho J., Wang B., Weddell G., Loring J., Kurahara C., Dudley E.C., Hayday A., Terhorst C., and Huang M. Abnormal T cell development in CD3-zeta-/- mutant mice and identification of a novel T cell population in the intestine EMBO J. 1993 12 12 4863 -4875
Love P.E. and Hayes S.M. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb Perspect. Biol. 2010 2 6 a002485
Love P.E., Shores E.W., Johnson M.D., Tremblay M.L., Lee E.J., Grinberg A., Huang S.P., Singer A., and Westphal H. T cell development in mice that lack the zeta chain of the T cell antigen receptor complex Science. 1993 261 5123 918 -921
Luton F., Buferne M., Legendre V., Chauvet E., Boyer C., and Schmitt-Verhulst A.M. Role of CD3gamma and CD3delta cytoplasmic domains in cytolytic T lymphocyte functions and TCR/CD3 down-modulation J. Immunol. 1997 158 9 4162 -4170
Malissen B., Ardouin L., Lyn S.Y., Guillet A., and Malissen M. Function of the CD3 subunits of the pre-TCR and TCR complexes during T cell development Adv. Immunol. 1999 72 103 -148
Malissen M., Gillet A., Ardouin L., Bouvier G., Trucy J., Ferrier P., Vivier E., and Malissen B. Altered T cell development in mice with a targeted mutation of the CD3-epsilon gene Embo J. 1995 14 19 4641 -4653
Malissen M., Gillet A., Rocha B., Trucy J., Vivier E., Boyer C., Kontgen F., Brun N., Mazza G., Spanopoulou E., Guy-Grand D., and Malissen B. T cell development in mice lacking the CD3-zeta/eta gene EMBO J. 1993 12 11 4347 -4355
Marcus N., Takada H., Law J., Cowan M.J., Gil J., Regueiro J.R., Plaza Lopez de Sabando D., Lopez-Granados E., Dalal J., Friedrich W., Manfred H., Hanson I.C., Grunebaum E., Shearer W.T., and Roifman C.M. Hematopoietic stem cell transplantation for CD3delta deficiency J. Allergy Clin. Immunol. 2011 128 5 1050 -1057
Martin E.P., Arnaud J., Alibaud L., Gouaillard C., Llobera R., Huchenq-Champagne A., and Rubin B. Molecular mechanisms in the TCR (TCR alpha beta-CD3 delta epsilon, gamma epsilon) interaction with zeta 2 homodimers: clues from a ‘phenotypic revertant’ clone Int. Immunol. 1999 11 7 1005 -1015
Mazariegos M.S., Muñoz-Ruiz M., Reiné J., Garcillán B., Recio M.J., Fernández-Malavé E., and Regueiro J.R. Inmunodeficiencias congénitas del receptor de antígeno de los linfocitos T Inmunología. 2013 32 3 94 -101
Mombaerts P., Clarke A.R., Rudnicki M.A., Iacomini J., Itohara S., Lafaille J.J., Wang L., Ichikawa Y., Jaenisch R., Hooper M.L., and Tonegawa S. Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages Nature. 1992 360 6401 225 -231
Morgan N.V., Goddard S., Cardno T.S., McDonald D., Rahman F., Barge D., Ciupek A., Straatman-Iwanowska A., Pasha S., Guckian M., Anderson G., Huissoon A., Cant A., Tate W.P., Hambleton S., and Maher E.R. Mutation in the TCRalpha subunit constant gene (TRAC) leads to a human immunodeficiency disorder characterized by a lack of TCRalphabeta+ T cells J. Clin. Invest. 2011 121 2 695 -702
Muñoz-Ruiz M., Perez-Flores V., Garcillán B., Guardo A.C., Mazariegos M.S., Takada H., Allende L.M., Kilic S.S., Sanal O., Roifman C.M., Lopez-Granados E., Recio M.J., Martinez-Naves E., Fernandez-Malave E., and Regueiro J.R. Human CD3γ, but not CD3δ, haploinsufficiency differentially impairs γδ versus αβ surface TCR expression BMC Immunol. 2013 14 1 3
Ozgur T.T., Asal G.T., Cetinkaya D., Orhan D., Kilic S.S., Usta Y., Ozen H., and Tezcan I. Hematopoietic stem cell transplantation in a CD3 gamma-deficient infant with inflammatory bowel disease Pediatr. Transpl. 2008 12 8 910 -913
Pacheco-Castro A., Alvarez-Zapata D., Serrano-Torres P., and Regueiro J.R. Signaling through a CD3 gamma-deficient TCR/CD3 complex in immortalized mature CD4+ and CD8+ T lymphocytes J. Immunol. 1998 161 6 3152 -3160
Perez-Aciego P., Alarcon B., Arnaiz-Villena A., Terhorst C., Timon M., Segurado O.G., and Regueiro J.R. Expression and function of a variant T Cell Receptor complex lacking CD3g J. Exp. Med. 1991 174 319 -326
Piirilä, H., Väliaho, J., and Vihinen, M. 2006. Available from http://structure.bmc.lu.se/idbase.
Recio M.J., Moreno-Pelayo M.A., Kilic S.S., Guardo A.C., Sanal O., Allende L.M., Perez-Flores V., Mencia A., Modamio-Hoybjor S., Seoane E., and Regueiro J.R. Differential biological role of CD3 chains revealed by human immunodeficiencies J. Immunol. USA. 2007 2556 -2564
Regueiro J.R., Arnaiz-Villena A., Ortiz de Landazuri M., Martin Villa J.M., Vicario J.L., Pascual-Ruiz V., Guerra-Garcia F., Alcami J., Lopez-Botet M., and Manzanares J. Familial defect of CD3 (T3) expression by T cells associated with rare gut epithelial cell autoantibodies Lancet. 1986 1 8492 1274 -1275
Reine J., Busto E.M., Muñoz-Ruiz M., Rossi N.E., Rodriguez-Fernandez J.L., Martinez-Naves E., Regueiro J.R., and Recio M.J. CD3gamma-independent pathways in TCR-mediated signaling in mature T and iNKT lymphocytes Cell Immunol. 2011 271 1 62 -66
Rieux-Laucat F., Hivroz C., Lim A., Mateo V., Pellier I., Selz F., Fischer A., and Le Deist F. Inherited and somatic CD3zeta mutations in a patient with T-cell deficiency N. Engl. J. Med. 2006 354 18 1913 -1921
Roberts J.L., Lauritsen J.P., Cooney M., Parrott R.E., Sajaroff E.O., Win C.M., Keller M.D., Carpenter J.H., Carabana J., Krangel M.S., Sarzotti M., Zhong X.P., Wiest D.L., and Buckley R.H. T-B+NK+ severe combined immunodeficiency caused by complete deficiency of the CD3zeta subunit of the T-cell antigen receptor complex Blood. 2007 109 8 3198 -3206
Saito T., Weiss A., Miller J., Norcross M.A., and Germain R.N. Specific antigen-Ia activation of transfected human T cells expressing murine Ti alpha beta-human T3 receptor complexes Nature. 1987 325 7000 125 -130
Sanal O., Yel L., Ersoy F., Tezcan I., and Berkel A.I. Low expression of T-cell receptor-CD3 complex: a case with a clinical presentation resembling humoral immunodeficiency Turk. J. Pediatr. 1996 38 1 81 -84
Sarantopoulos S., Stevenson K.E., Kim H.T., Cutler C.S., Bhuiya N.S., Schowalter M., Ho V.T., Alyea E.P., Koreth J., Blazar B.R., Soiffer R.J., Antin J.H., and Ritz J. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease Blood. 2009 113 16 3865 -3874
Siegers G.M., Swamy M., Fernandez-Malave E., Minguet S., Rathmann S., Guardo A.C., Perez-Flores V., Regueiro J.R., Alarcon B., Fisch P., and Schamel W.W. Different composition of the human and the mouse gammadelta T cell receptor explains different phenotypes of CD3gamma and CD3delta immunodeficiencies J. Exp. Med. USA. 2007 2537 -2544
Soudais C., de Villartay J.P., Le Deist F., Fischer A., and Lisowska-Grospierre B. Independent mutations of the human CD3-epsilon gene resulting in a T cell receptor/CD3 complex immunodeficiency Nat. Genet. 1993 3 1 77 -81
Sussman J.J., Bonifacino J.S., Lippincott-Schwartz J., Weissman A.M., Saito T., Klausner R.D., and Ashwell J.D. Failure to synthesize the T cell CD3-zeta chain: structure and function of a partial T cell receptor complex Cell. 1988 52 1 85 -95
Szymczak A.L., Workman C.J., Wang Y., Vignali K.M., Dilioglou S., Vanin E.F., and Vignali D.A. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector Nat. Biotechnol. 2004 22 5 589 -594
Takada H., Nomura A., Roifman C.M., and Hara T. Severe combined immunodeficiency caused by a splicing abnormality of the CD3delta gene Eur. J. Pediatr. 2005 164 5 311 -314
Thoenes G., Soudais C., le Deist F., Griscelli C., Fischer A., and Lisowska-Grospierre B. Structural analysis of low TCR-CD3 complex expression in T cells of an immunodeficient patient J. Biol. Chem. 1992 267 1 487 -493
Tokgoz H., Caliskan U., Keles S., Reisli I., Guiu I.S., and Morgan N.V. Variable presentation of primary immune deficiency: two cases with CD3 gamma deficiency presenting with only autoimmunity Pediatr. Allergy Immunol. 2013 24 3 257 -262
Torres P.S., Alcover A., Zapata D.A., Arnaud J., Pacheco A., Martin-Fernandez J.M., Villasevil E.M., Sanal O., and Regueiro J.R. TCR dynamics in human mature T lymphocytes lacking CD3 gamma J. Immunol. 2003 170 12 5947 -5955
Torres P.S., Zapata D.A., Pacheco-Castro A., Rodriguez-Fernandez J.L., Cabanas C., and Regueiro J.R. Contribution of CD3 gamma to TCR regulation and signaling in human mature T lymphocytes Int. Immunol. 2002 14 11 1357 -1367
Tsuzaka K., Fukuhara I., Setoyama Y., Yoshimoto K., Suzuki K., Abe T., and Takeuchi T. TCR zeta mRNA with an alternatively spliced 3’-untranslated region detected in systemic lupus erythematosus patients leads to the down-regulation of TCR zeta and TCR/CD3 complex J. Immunol. 2003 171 5 2496 -2503
Tsuzaka K., Setoyama Y., Yoshimoto K., Shiraishi K., Suzuki K., Abe T., and Takeuchi T. A splice variant of the TCR zeta mRNA lacking exon 7 leads to the down-regulation of TCR zeta, the TCR/CD3 complex, and IL-2 production in systemic lupus erythematosus T cells J. Immunol. 2005 174 6 3518 -3525
Wada T. and Candotti F. Somatic mosaicism in primary immune deficiencies Curr. Opin. Allergy Clin. Immunol. 2008 8 6 510 -514
Wang Y., Becker D., Vass T., White J., Marrack P., and Kappler J.W. A conserved CXXC motif in CD3epsilon is critical for T cell development and TCR signaling PLoS Biol. 2009 7 12 e1000253
Wegener A.M., Hou X., Dietrich J., and Geisler C. Distinct domains of the CD3-gamma chain are involved in surface expression and function of the T cell antigen receptor J. Biol. Chem. 1995 270 9 4675 -4680
Weiss A. and Stobo J.D. Requirement for the coexpression of T3 and the T cell antigen receptor on a malignant human T cell line J. Exp. Med. 1984 160 5 1284 -1299
Zapata D.A., Schamel W.W., Torres P.S., Alarcon B., Rossi N.E., Navarro M.N., Toribio M.L., and Regueiro J.R. Biochemical differences in the alphabeta T cell receptor.CD3 surface complex between CD8+ and CD4+ human mature T lymphocytes J. Biol. Chem. 2004 279 23 24485 -24492
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 1 • March 2015
Pages: 3 - 19
History
Received: 27 August 2014
Accepted: 28 October 2014
Accepted manuscript online: 29 October 2014
Version of record online: 29 October 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Ana V.M. Marin, Beatriz Garcillán, Anaïs Jiménez-Reinoso, Miguel Muñoz-Ruiz, Alejandro C. Briones, Edgar Fernández-Malavé, Maria J. Recio, and José R. Regueiro. 2015. Human congenital T-cell receptor disorders. LymphoSign Journal.
2(1): 3-19. https://doi.org/10.14785/lpsn-2014-0012
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
