Monoallelic STAT1 mutations and disease patterns
Abstract
Monoallelic mutations in STAT1 are associated with a myriad of clinical phenotypes. Some clinical patterns appear to be preferentially associated with mutations in various STAT1 domains. Included are trends of phenotype-genotype correlations in patients with mutations in STAT1.
In the past several years it became apparent that mutations in STAT1 are the most common cause of chronic mucocutaneous candidiasis (CMCC) in North America.
The clinical features of thrush and skin and nail lesions are indistinguishable between CMCC caused by an autosomal recessive mutation in the AIRE gene, which was described 2 decades ago (Ahonen et al. 1990; Finnish-German APECED Consortium 1997), or the recently identified monoallelic mutations in STAT1 (van de Veerdonk et al. 2011) (Figure 1). Autoimmune manifestations including endocrinopathies are also shared between these genetic entities. However, it appears that although hypoparathyroidism and hypoadrenodism are more common in AIRE deficiency, hypothyroidism and diabetes mellitus are more common with STAT1 mutations. Moreover, recurrent respiratory infections and antibody deficiency have a confirmed association with STAT1 abnormalities, but only rarely in AIRE deficiency (Bentur et al. 1991).
Figure 1:
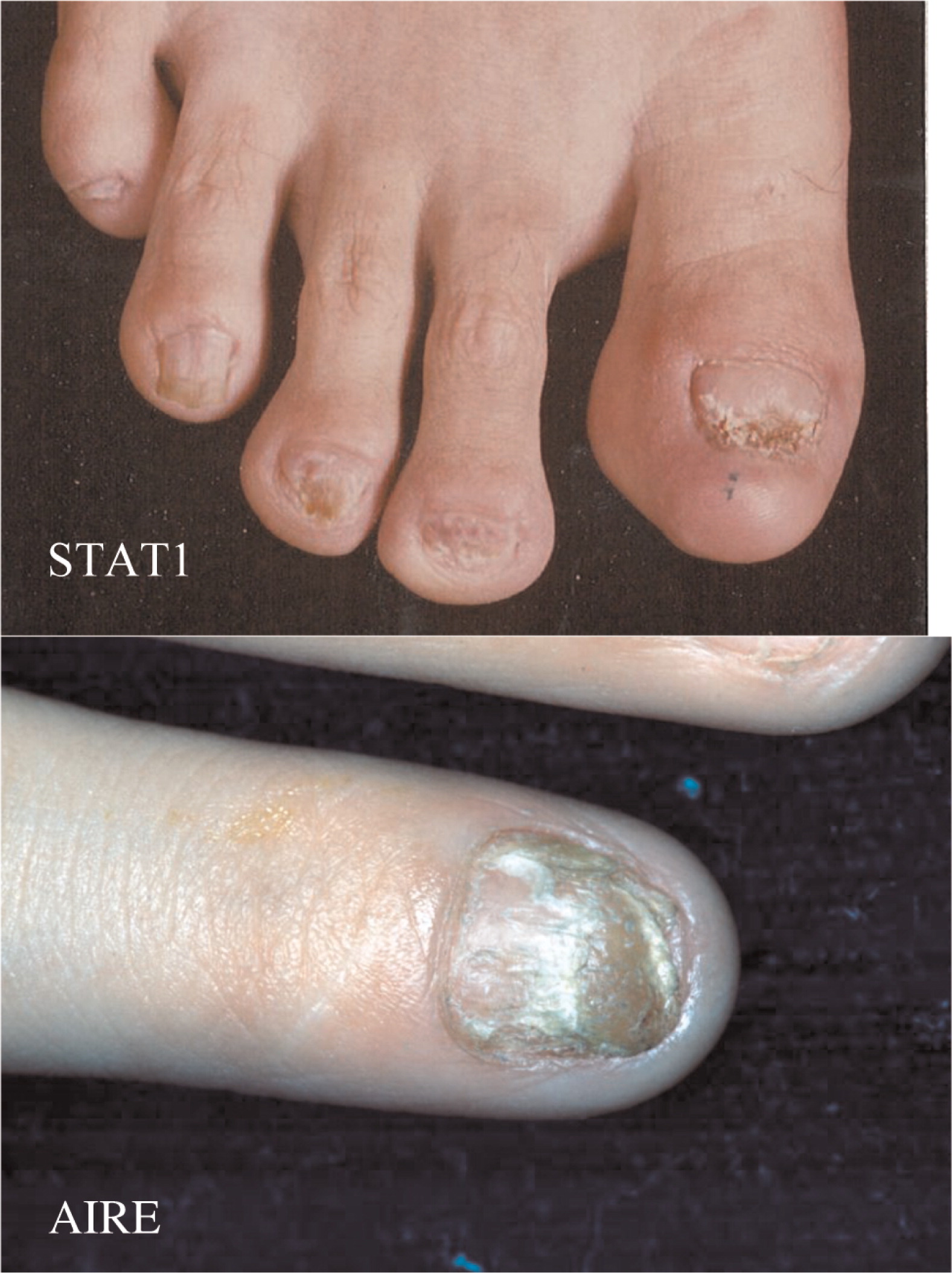
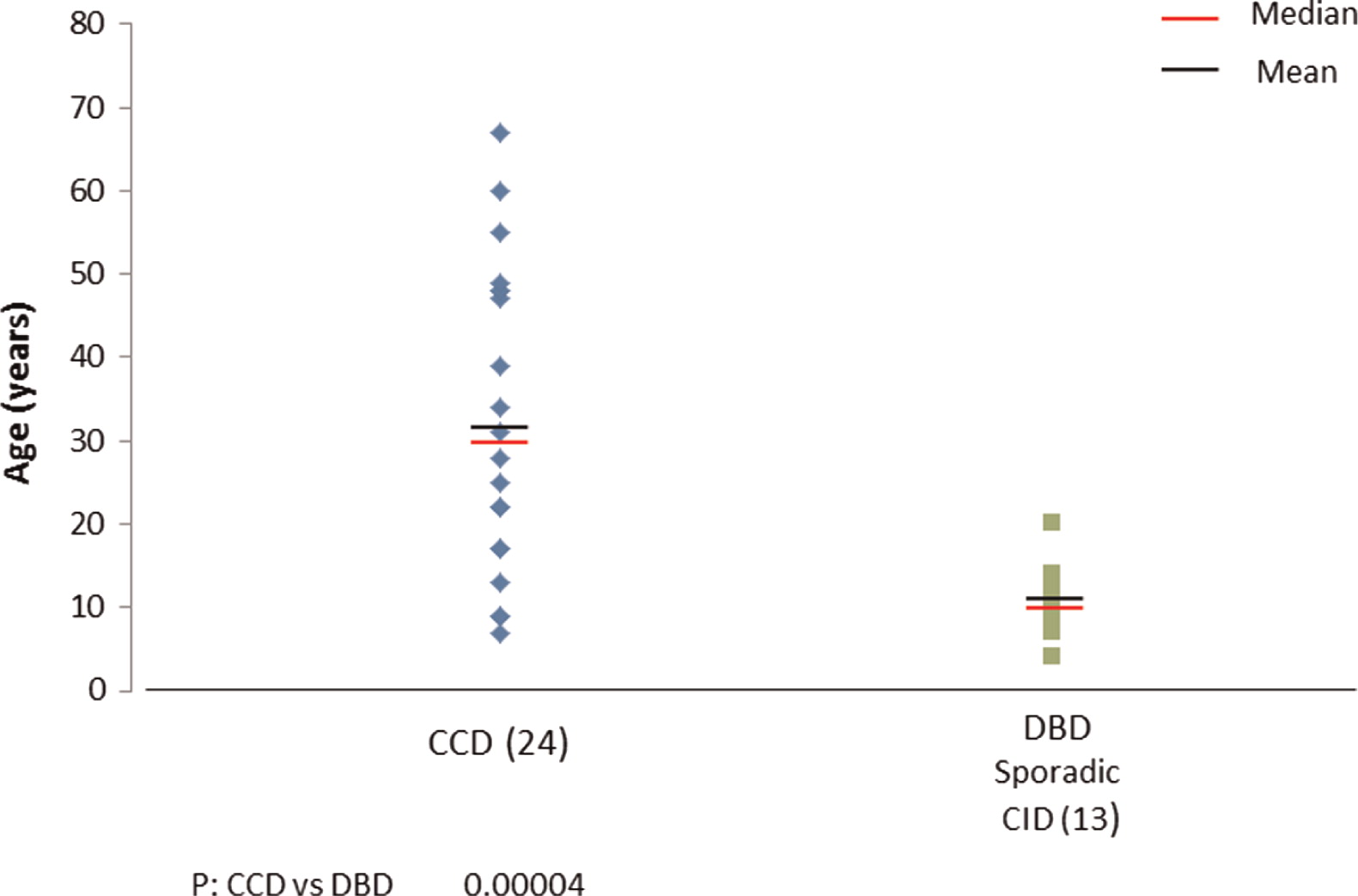
Interestingly, clinical entities formerly thought to be distinct, such as autosomal dominant CMCC, are also associated with mutations in STAT1. The clinical spectrum in these cases is wide (see article by Nahum and Dalal on page 97) ranging from vascular disease in the central nervous system to very mild symptoms of isolated oral thrush. Upon review of all published cases (Liu et al. 2011; Maródi et al. 2012; Takezaki et al. 2012; Sampaio et al. 2013; Uzel et al. 2013; Sharfe et al. 2014), it appears that mutations associated with autosomal dominant CMCC are mostly localized to the coiled–coil domain of STAT1. The mean and median ages of both of these patients at the time of reporting appears to be around 30 years of age (Figure 2), but more striking are the mean and median ages of death in this group of patients as young as 35 years and 28 years, respectively (Figure 3). The cause of death was squamous cell carcinoma in the older patients and stroke in younger individuals.
Figure 2:
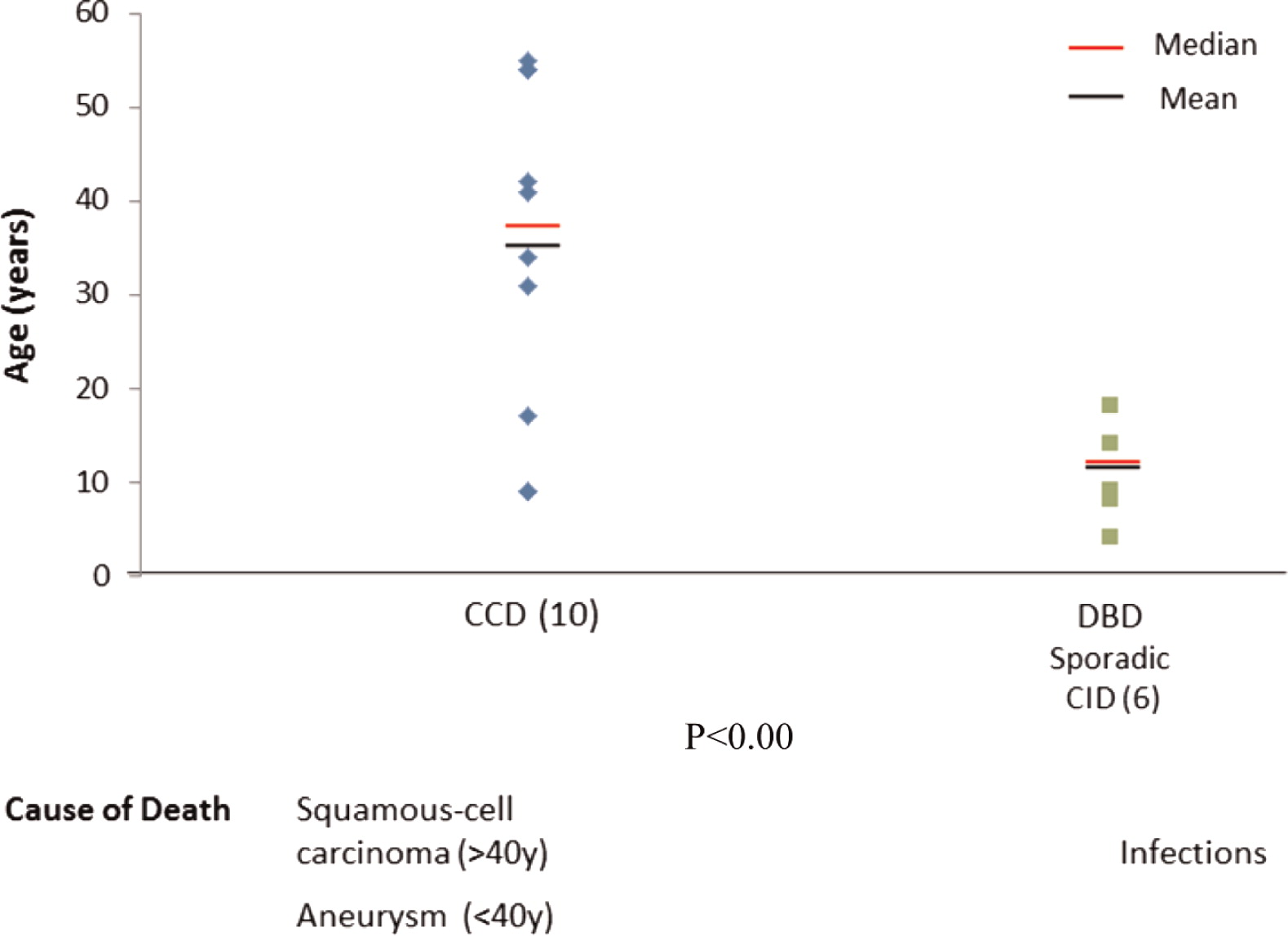
Figure 3:
Recently, whole exome sequencing discovered a surprising phenotype of monoallelic STAT1 deficiency (Sharfe et al. 2014). Invariably, this group of patients had no family history of immunodeficiency. They present early in life with repeated viral and bacterial infections, and they suffer chronic colitis as well as other autoimmune manifestations. Evaluation of their immune system reveals a progressive decline in immunity, culminating in a combined immunodeficiency including profound lymphopenia and antibody deficiency, practically developing combined immunodeficiency (CID). Most of these patients have mutations in the DNA binding domain (DBD) of STAT1.
These patients with sporadic CID invariably die of overwhelming viral infection at a mean and median age of 10 years. This analysis, albeit imperfect, suggests clustering of phenotype–genotype correlation.
To date there is no cure for STAT1-related disorders. Although control of bacterial infections can be achieved by using antibiotic therapy and immunoglobulin replacement, other manifestations cannot be prevented or effectively treated. Attempts to clear chronic cytomegalovirus, Epstein–Barr virus, or John Cunningham virus were not successful in these patients. Moreover, vascular changes to the central nervous system, including vasculitis, aneurisms, and acute bleeding cannot be effectively and completely prevented, even with harsh immunosuppressive treatments with cyclophosphoramide. The use of high dose intravenous immunoglobulin failed to show a significant improvement, but the use of immune modulation agents such as Mycophenolate Mofetil may be of benefit in controlling some autoimmune features.
For cases with mutations in the DBD domain, especially those known to be associated with mortality at an early age, such as T385M, hemotopoietic stem cell therapy might be a plausible consideration.
REFERENCES
Ahonen P., Myllärniemi S., Sipilä I., and Perheentupa J. Clinical variation of autoimmune polynendocrinolopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients N. Engl. J. Med. 1990 322 1829
Bentur L., Nisbet-Brown E., Levison H., and Roifman C.M. Lung disease associated with IgG subclass deficiency in chronic mucocutaneous candidiasis J. Pediatr. 1991 118 82
Finnish–German APECED Cosortium An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains Nat. Genet. 1997 17 399
Liu L., Okada S., Kong X.F., Smeekens S.P., Joosten L.A.B., Gilissen C., Arts P., Rosentul D.C., Carmichael A.J., Smits-van der Graaf C.A.A., Bart Jan Kullberg B.J., van der Meer J.W.M., Desa Lilic D., Veltman J.A., and Netea M.G. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis J. Exp. Med. 2011 208 1635
Maródi L., Cypopwyj S., Tóth B., Smeekens S.P., Joosten L.A.B., Gilissen C., Arts P., Rosentul D.C., Carmichael A.J., Smits-van der Graaf C.A.A., Bart Jan Kullberg B.J., van der Meer J.W.M., Desa Lilic D., Veltman J.A., and Netea M.G. Molecular mechanisms of mucocutaneous immunity against cadida and Staphylococcus species J. Allergy. Clin. Immunol. 2012 130 1019
Sampaio E.P., Hsu A.P., Pechacek J., Bax H.I., Dias D.L., Paulson M.L., Chandrasekaran P., Rosen L.B., Carvalho D.S., Ding L., Vinh D.C., Browne S.K., Datta S., Milner J.D., Kuhns D.B., Long Priel D.A., Sadat M.A., Shiloh M., De Marco B., Alvares M., Gillman J.W., Ramarathnam V., de la Morena M., Bezrodnik L., Moreira I., Uzel G., Johnson D., Spalding C., Zerbe C.S., Wiley H., Greenberg D.E., Hoover S.E., Rosenzweig S.D., Galgiani J.N., and Holland S.M. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation and disseminated coccidiodomycosis and histoplasmosis J. Allergy. Clin. Immunol. 2013 131 1624
Sharfe N., Nahum A., Newell A., Dadi H., Ngan B., Pereira S.L., Herbrick J.A., and Roifman C.M. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1 J. Allergy. Clin. Immunol. 2014 133 807
Takezaki S., Yamada M., Kato M., Park M.J., Maruyama K., Yamazaki Y., Chida N., Ohara O., Kobayashi I., and Ariga T. Chronic Mucocutaneous candidiasis caused by a gain-of-function mutation in the STAT1 DNA-binding domain J. Immunol. 2012 189 1521
Uzel G., Sampaio E.P., Lawrence M.G., Hsu A.P., Hackett M., Dorsey M.J., Noel R.J., Verbsky J.W., Freeman A.F., Janssen E., Bonilla F.A., Pechacek J., Chandrasekaran P., Browne S.K., Agharahimi A., Gharib A.M., Mannurita S.C., Yim J.J., Gambineri E., Torgerson T., Tran D.Q., Milner J.D., and Holland S.M. Dominant gain-of-funtion STAT1 mutationsin FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome J. Allergy. Clin. Immunol. 2013 131 1611
Van de Veerdonk F.L., Plantinga T.S., Hoischen A., Smeekens S.P., Joosten L.A.B., Gilissen C., Arts P., Rosentul D.C., Carmichael A.J., Smits-van der Graaf C.A.A., Bart Jan Kullberg B.J., van der Meer J.W.M., Desa Lilic D., Veltman J.A., and Netea M.G. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis N. Eng. J. Med. 2011 365 54
Chaim M. Roifman, MD, FRCP, FCACB
Editor-in-Chief
Information & Authors
Information
Published In

LymphoSign Journal
Volume 1 • Number 2 • December 2014
Pages: 57 - 59
History
Accepted manuscript online: 11 September 2014
Version of record online: 11 September 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
RoifmanChaim M.MD, FRCP, FCACB. 2014. Monoallelic STAT1 mutations and disease patterns. LymphoSign Journal.
1(2): 57-59. https://doi.org/10.14785/lpsn-2014-0011
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. The effects of STAT1 dysfunction on the gut
2. Autosomal dominant transmission of signal transduction and activator of transcription 1 (STAT1) mutation (Thr385Met) and extended lifespan
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
