Homozygous NF-kB1 mutation causing combined immunodeficiency: a histopathological analysis
Abstract
Introduction: The nuclear factor-kB (NF-kB) signaling pathway plays a major role in mediating multiple cellular processes, including immune and inflammatory responses.
Aims: We describe the histopathological findings of lymph nodes from a patient with a homozygous NF-kB subunit 1 (NF-kB1) mutation causing a combined immunodeficiency phenotype.
Results: A nodal biopsy was performed for lymphadenopathy evaluation, in the context of development of persistent EBV infection. Our findings show that this patient has normal lymph node tissue present, however, abnormal histopathology features were observed, including atrophic germinal centers. B cell subset components within the B cell domain were also analyzed. The development of the B cell response during EBV infection was found to be significantly impaired.
Conclusion: Aberrant signaling due to NF-kB1 deficiency has a significant impact on the development of B cell immunoproliferative responses.
Statement of novelty: We report on the abnormal histopathology findings of lymph node biopsy from a patient with homozygous NF-kB1 mutation.
Background
The nuclear factor-kB (NF-kB) signaling pathway plays a major role in mediating multiple cellular processes, including immune and inflammatory responses, lymphocyte development, ectodermal development, cell growth, and programmed death (Zhang et al. 2017). The NF-kB transcription factor family consists of 5 Rel proteins, p50/p105, p52/p100, RelA, RelB, and c-Rel, which dimerize with each other and drive or inhibit gene expression in the nucleus. There are 2 well characterized NF-κB signaling pathways, the canonical and non-canonical pathways (Gilmore 2006).
Recently, a novel homozygous mutation in the NF-κB subunit 1 (NF-kB1) gene, encoding p50/p105, was described as causing a profound disruption of the classical NF-kB pathway resulting in combined immunodeficiency (CID). The mutation in NFKB1 predicted a glycine to arginine substitution at position 960 (p.Gly960Arg). G960R is located in the C-terminal of p105, distal to the death domain. This position is highly conserved across vertebrates and thus a change is expected to have significant pathogenicity which manifests with abnormal differentiation of T and B cells, aberrant specific antibody formation, cytokine secretion, and cell proliferation (Mandola et al. 2021).
In addition to immunological investigation and genetic analysis, histopathological examination of tissue may have an important role in supporting the diagnosis of immunodeficiency or to be perused if malignancy is suspected as a complication. As a consequence of inborn errors in immunity, variable defects of lymphocyte development and morphology of lymph organs may occur. The lack of NF-κB1 appears to also markedly disrupt the architecture of lymphoid tissues as was shown in NF-κB1-/- mice (Weih et al. 2001; Lo et al. 2006).
There are several known findings on lymph node histopathology in SCID that vary according to underlying pathophysiology. It has been reported that lymph nodes from T-B+ SCID patients regularly contain B cells, either as scattered cells or remnants of follicles without formation of germinal centers. T-B- SCID patients have severe depletion of B and T cells, and resulting “stromal” nodes contain fibroblasts, flat endothelial cells, and macrophages. In SCID due to JAK3 deficiency the lymphoid tissue is completely absent (Ratech et al. 1989; Facchetti et al. 1998).
In this study, we describe the histopathological features within the lymph node of a patient with loss of a major B cell activation pathway initiator, due to the presence of a homozygous mutation in NF-kB1. The lymph node was biopsied and examined when the patient developed persistent lymphadenopathy due to EBV infection. Our findings demonstrate that the loss of NF-kB1 did not affect the development or histogenesis of B or T cells, however, there was a major impact on the development of B cell immuno proliferative responses within the germinal centers.
Case presentation
Our patient, a 7 year old male born to consanguineous parents of Pakistani-descent, was recently diagnosed with a novel homozygous G to A transition (c.2878G>A) in the NFKB1 gene (Mandola et al. 2021).
His clinical history was significant for severe, flaky, and difficult to manage atopic dermatitis, repeated episodes of otitis media by the age of 3 months, chronic diarrhea at 5 months, failure to thrive, and lymphadenopathy. At the age of 7 months, he was hospitalized for respiratory distress syndrome, requiring assisted ventilation secondary to Pneumocystis jirovecii. On that hospital admission he also developed candidial urinary tract infection (UTI). At age 9 months, he was admitted for prolonged fever lasting 2 weeks, diarrhea, generalized lymphadenopathy, hepatosplenomegaly, and increased liver enzymes in association with very high copy numbers of EBV (104 to 106 copies) detected in the blood and on bronchoalveolar lavage (BAL). On that admission he was also treated for enterococcal UTI. Despite regular intravenous immunoglobulin replacement and periodic treatment with ganciclovir, he continued to experience repeated episodes of microbial infections as well as periodic increase in EBV viremia associated with generalized lymphadenopathy.
Laboratory workup revealed elevated IgE and eosinophil levels. His further work-up showed normal IgG, elevated IgA and low IgM levels. He was also found to have an abnormal humoral immune response to vaccinations. Total lymphocyte and T cell counts were normal, but over time, a gradual reduction in CD4+ cells were observed and the number of circulating CD8+ T cells increased over time. T cell depressed responsiveness to mitogen stimulation was noted and T cell receptor repertoire analysis appeared skewed. Analysis of CD4+ cells demonstrated under representation of several Vβ families, including Vβ11, Vβ13.6, and Vβ20, and overrepresentation of Vβ4, Vβ7.2, Vβ13.1, and Vβ18. CD8+ T cells showed under representation of Vβ5.3 and Vβ7.1, however, overrepresentation of Vβ2, Vβ3, Vβ5.2, Vβ7.2, Vβ14, and Vβ16 was prominent, suggesting peripheral T-cell expansion, and in complete agreement with the marked shift to differentiated cells observed in flow cytometry studies.
Outcome and follow up
Our patient has been relatively well and is currently on Pneumocystis jirovecii prophylaxis and intravenous immunoglobulin replacement therapy. Human leukocyte antigen evaluation has not been suggestive of a compatible match in the family for hematopoietic stem cell transplantation, and given his age and infectious complications in the past, his course of transplant may be complicated. Given that his past few years have been uneventful together with the considerations mentioned, hematopoietic stem cell transplantation has not become a part of his treatment.
Methods
Patient
Patient informed consent was obtained, and clinical information was collected from medical records in accordance with approved protocols from the Research Ethics Board at the Hospital for Sick Children.
Lymph node histopathology
The nodal biopsy was performed during the initial clinical evaluation of this patient, in the context of development of persistent EBV infection and was obtained from the left inguinal region.
Lymph node was received fresh, fixed in buffered formalin for 24 hours and subsequently embedded in paraffin. Control lymph nodes were selected from the tissue archive from patients diagnosed with non-specific reactive lymphadenopathies. Histologic sections of the patient’s lymph node were stained with hematoxylin eosin (H&E) or for immunohistological studies, unstained sections were stained with the following commercially available antibodies supplied by DAKO/Agilent (Santa Clara, California, US) : pan-T cell receptor CD3 (rabbit polyclonal GA503), germinal B cell marker Bcl-6 (monoclonal Clone BG-B6P), activated post germinal center B cell/plasma cell marker Mum-1 (MUltiple Myeloma 1), and anti-apoptosis protein Bcl-2 (monoclonal Clone 124). Antibodies were supplied pre-diluted and the de-paraffinized slides were incubated with each of the antibodies according to the staining procedures supplied by the supplier.
The post-antibody binding detection was amplified using the polymer amplification and visualized by using Di-azo-benzidine (DAB) as a chromogen. All of these were suppled as Envision Flex High pH kit) Stains were performed using an automated staining system by DAKO Omnis.
Bcl-6 and Mum-1 immunostaining were used as germinal center markers for histological characterization and malignancy detection.
Results
Lymph node pathology
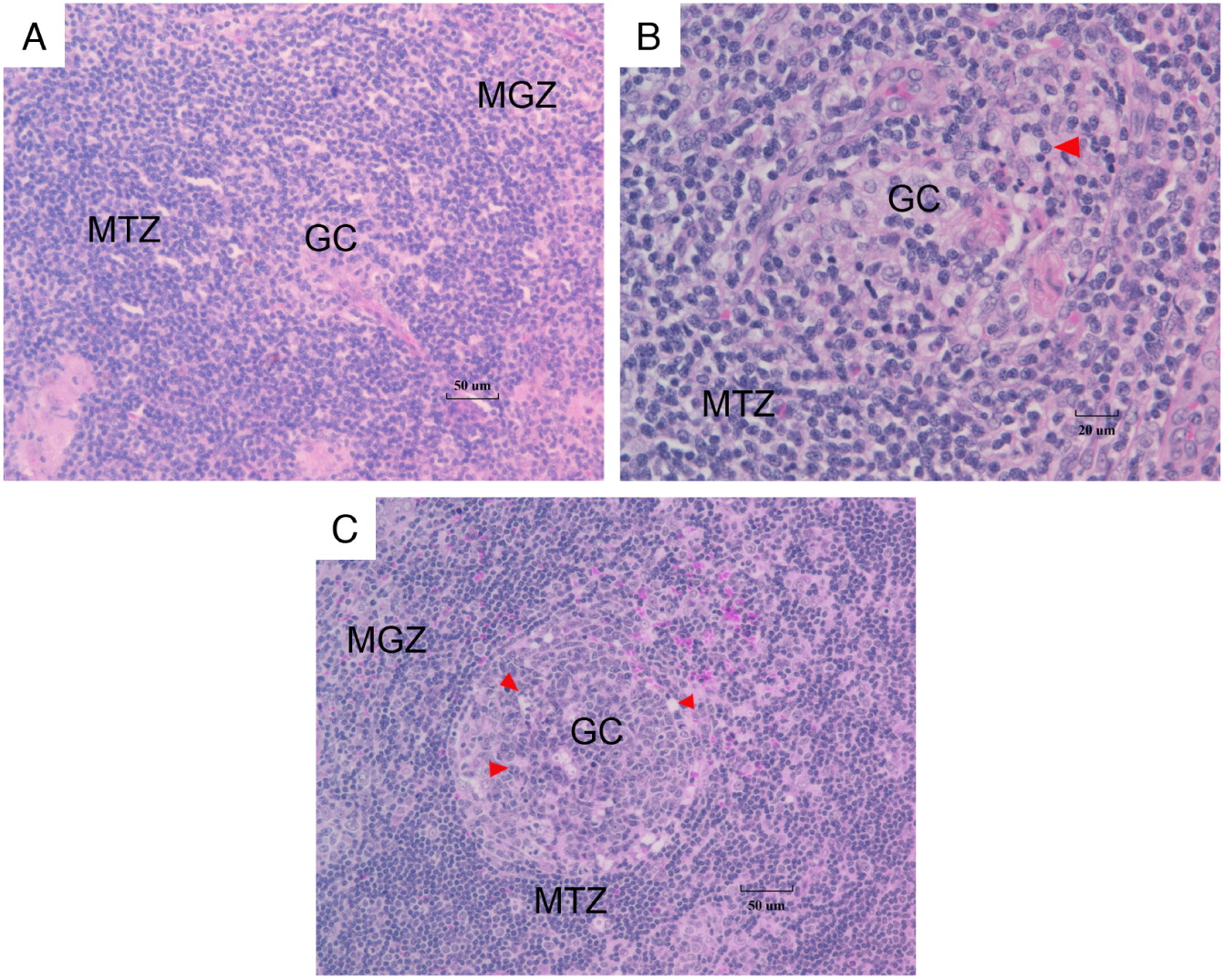
Abnormal histopathology features in the lymph node of this patient were observed. H&E stained sections showed atrophic/underdeveloped germinal centers throughout the lymph node cortex (Figures 1A and 1B) compared to control (Figure 1C). Many had the appearance of primary germinal centers (Figure 1A), surrounded by a thick cuff of mantle cells. Nonreactive marginal zones were noted. There was a lack of reactive features within the germinal center domain (Figure 1B), including no tingible body macrophages and only small numbers of centroblasts present. There is no segregation of the centroblasts and centrocytes into pale and dense zones.
Figure 1:
Using a combination of immunostains (including the pan B cell marker CD20, follicular dendritic cell marker CD21, and T cell receptor complex antigen CD3), the immune-anatomical locations and development of B cell, T cell, and follicular dendritic cell domains within the patient’s lymph node were found to be normal (not shown). The B cell subset components within the B cell domain were analyzed further with a panel of antibodies to detect proteins that relate to B cell maturation and function.
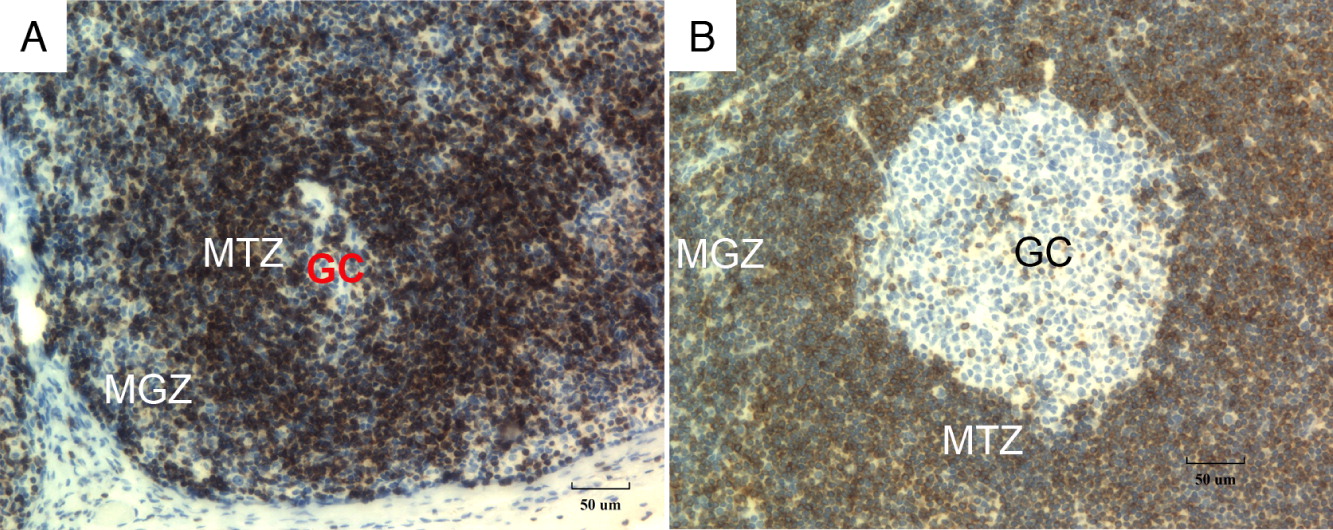
Bcl-2 protein, which blocks apoptosis, is normally absent within the reactive centrocytes and centroblasts within the germinal center and is expressed on the mantle B lymphocytes and adjacent T cells. In normal lymph nodes, immunostaining for Bcl-2 revealed germinal centers that were significantly diminished in size (Figures 2A and 2B) compared to control (Figure 2C).
Figure 2:
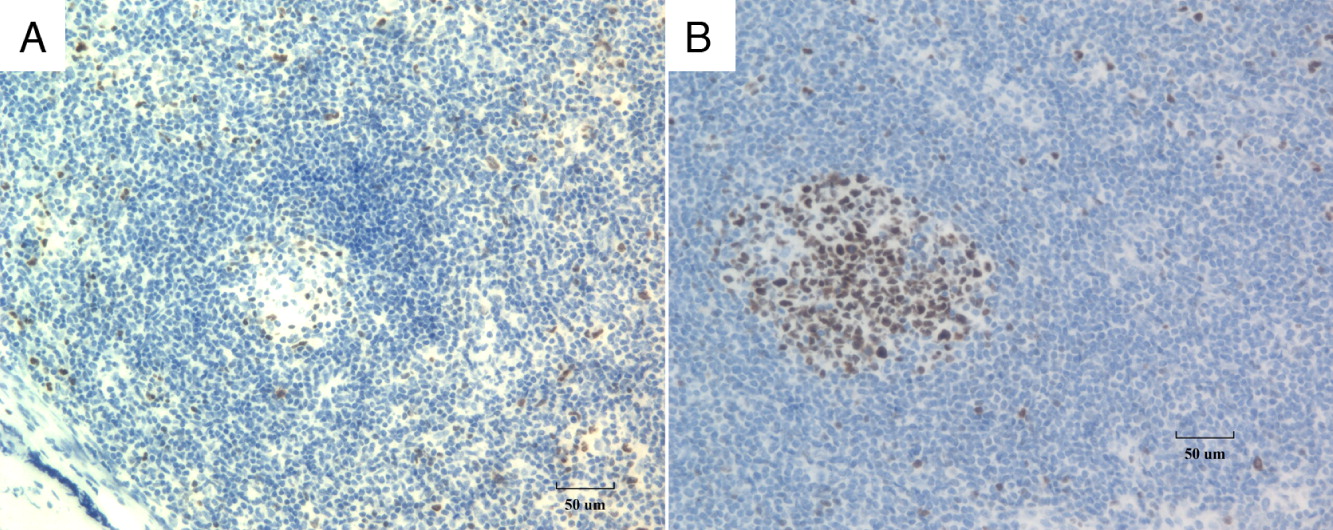
Bcl-6 is a transcriptional repressor required in mature B cells during the germinal center reaction, through DNA binding and recruitment of co-repressor complexes on the promoter of its targets. Its expression is tightly regulated during mature B cell differentiation, with Bcl-6 protein expression restricted to the germinal center stage of differentiation (Basso and Dalla-Favera 2012). Bcl-6 is critical for the development of a diverse primary B cell repertoire and protects germinal center B cells against DNA damage-induced apoptosis during somatic hypermutation and class switch recombination (Duy et al. 2010). Histologically, Bcl-6 is expressed by B cells that migrate into germinal centers and is thus a useful marker for germinal center B cells. In our patient, immunostaining for Bcl-6 (Figure 3A) demonstrated almost complete loss of Bcl-6 positive cells in the germinal centers while some positively stained cells could be detected in the peripheral areas. This is in contrast to the normal control (Figure 3B), where the majority of Bcl-6 cells are present in the germinal center. Together, this suggests that without proper NF-κB1 signaling, these cells fail to migrate to the germinal centers, or alternatively, they fail to transform into activated B cells and continue to emigrate bearing the Bcl-6 marker.
Figure 3:
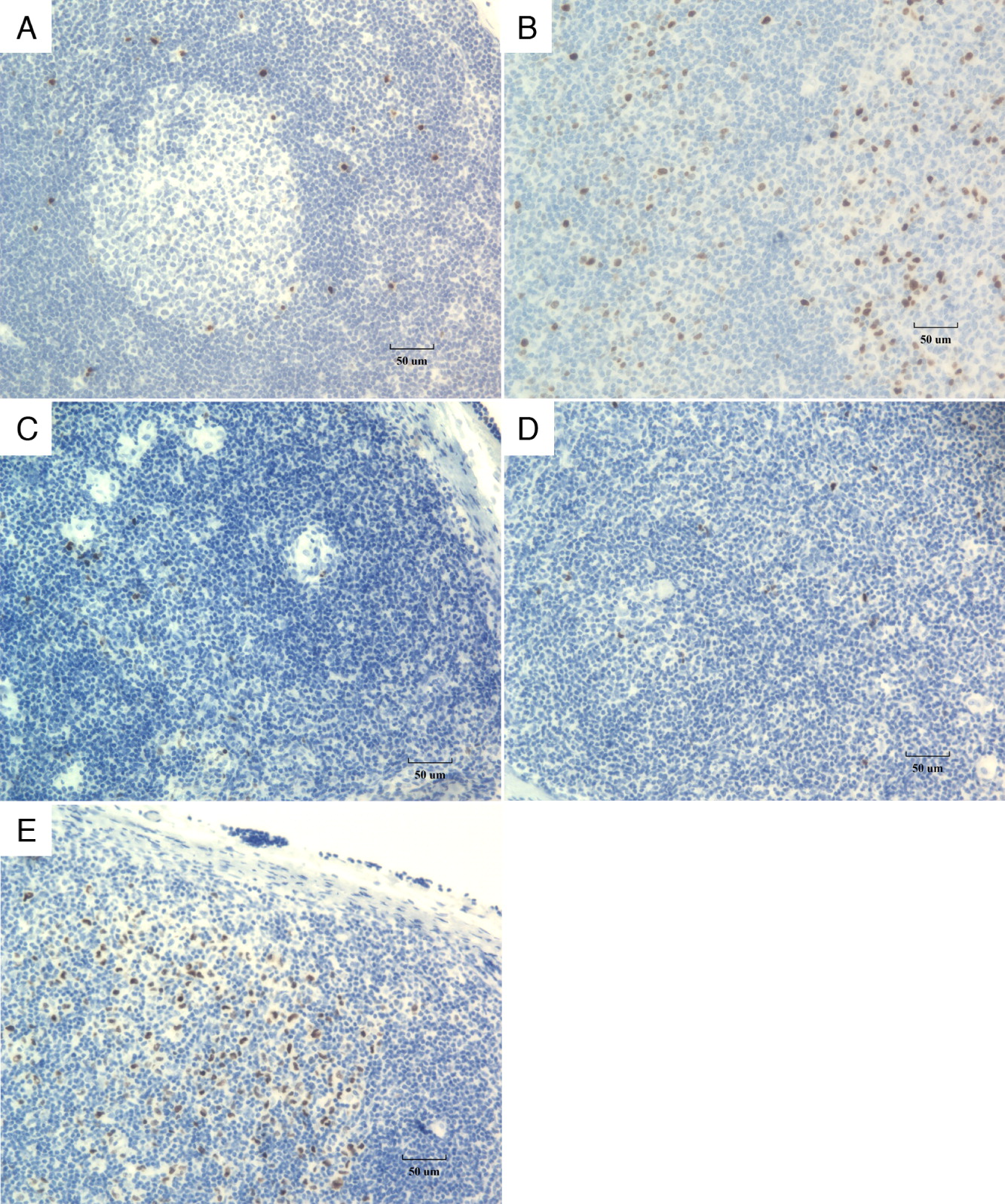
Mum-1 is a transcriptional factor which is expressed in the final step of intra-germinal center B cell differentiation and in post-germinal center (late centrocyte) or activated B cells (Gualco et al. 2010). It is normally expressed in the nuclei and cytoplasm of plasma cells and a small percentage of germinal center B cells mainly located in the “light zone”. Polymerase chain reaction (PCR) analysis of Mum-1 positive cells isolated from germinal centers have previously shown that they contain rearranged Ig heavy chain genes with a varying number of somatic mutations (Falini et al. 2000). In normal lymph nodes, Mum-1 immunostaining demonstrates that post-germinal center B cells gather in the interfollicular areas, including parafollicular sinusoids (Figures 4A and 4B). In our NF-kB1 deficient patient, immunostaining revealed a marked reduction in numbers of Mum-1-positive activated B cells in the germinal centers and the interfollicular zones (Figure 4C). The reduced development of secondary germinal centers (Figure 4D) indicates that the development of activated B cells is likely slowed although not completely absent, given the presence of Mum-1 positive cells in the lymph node sinusoids (Figure 4E).
Figure 4:
Discussion
Multiple studies have described NFKB1 mutations as a monogenic cause of immunodeficiency, mostly presenting in late childhood/early adulthood. Patients with NFKB1 haploinsufficiency have a variety of clinical manifestations, including predominantly antibody deficiency, autoimmunity, and immune dysregulation. Autoinflammatory disorders including Bechet disease have been reported as well (Smart 2014; Kaustio et al. 2017; Tuijnenburg et al. 2018; Lorenzini et al. 2020).
Recently, a young pediatric patient who first presented with severe dermatitis, recurrent invasive and opportunistic infections, persistent EBV viremia, generalized lymphadenopathy and hepatosplenomegaly at the age of 9 months was diagnosed with a novel homozygous mutation in the NFKB1 gene. This mutation predicted to cause a glycine to arginine substitution at position 960 (p.Gly960Arg). G960R is located in the C-terminal of p105, distal to the death domain and the Ser-903 and Ser-907 phosphorylation sites required for p105 proteolytic processing. Phosphorylation of p105 directly affects the formation of p50 which is required for downstream transcription, i.e. attenuated NF-kB1 phosphorylation on stimulation leads to reduced nuclear RelA translocation and target gene expression (Mandola et al. 2021). To our knowledge, this is the only case reported in the literature as causing a profound disruption of the classical NF-kB pathway resulting in SCID.
Lymph nodes are organized secondary lymphoid organs with a distinct architecture, containing structures comprising a reticular network packed with lymphocytes, macrophages, and dendritic cells. They are divided into microenvironments, such as lymphoid follicles and additional B and T cell areas, all surrounded by a fibrous capsule. A normal reactive lymph node histopathology is expected to consist of numerous follicles, prominent germinal centers, “tingible body macrophages”, i.e., prominent macrophages with irregular cellular debris. These macrophages process antigens to pass to lymphocytes to stimulate a specific immune response (Willard-Mack 2006).
Our patient has normal lymphoid tissue development as determined by CD20, CD21 and CD3 staining, however, histopathological findings revealed atrophic germinal centers, thick concentric mantle zones and nonreactive marginal zones. The areas within the germinal center where centrocytes and centroblasts reside were small, and there was a lack of tingible body macrophages in comparison to the normal control. Immunostains targeting the B cells identified B cell follicles that were small and lacked expansion of germinal center domains. Furthermore, B cells failed to mature and emigrate at a regular rate.
Together, the histopathological observations are in keeping with combined immunodeficiency and likely explain this patient’s inability to clear EBV infection as well as the very high levels of EBV copy number in the blood of the patient. Moreover, our findings may suggest that the failure to develop a normal nodal architecture is related to a particularly severe T cell defect and thereby impairment of the T-B cell interaction. Failure to develop normal germinal center structure of this patient is also an indication of the important role of the NF-κB1 pathway in normal lymphoid architecture development.
NF-κB1 gene knockout studies have demonstrated the critical role of NF-κB1 in the development of lymphoid tissue. NF-κB1-/- mice display markedly disrupted lymphoid tissue architecture with flat Payer patches and lack of clear follicular structures (Willard-Mack 2006). In our patient, while lymphoid tissue development was normal, the development of the B cell response during EBV infection was observed to be markedly hindered.
Conclusion
Although homozygous NF-kB1 mutation in our patient resulted in a normal lymphatic tissue development, the loss of NF-kB activation mechanism has a significant impact on the development of B cell immunoproliferative responses within the germinal centers.
REFERENCES
Basso K. and Dalla-Favera R. 2012. Roles of BCL6 in normal and transformed germinal center B cells. Immunol. Rev. 247(1): 172–183.
Duy C., Yu J.J., Nahar R., Swaminathan S., Kweon S.M., Polo J.M., Valls E., Klemm L., Shojaee S., Cerchietti L., Schuh W., Jäck H.M., Hurtz C., Ramezani-Rad P., Herzog S., Jumaa H., Koeffler H.P., de Alborán I.M., Melnick A.M., Ye B.H., and Müschen M. 2010. BCL6 is critical for the development of a diverse primary B cell repertoire. J. Exp. Med. 207(6): 1209–1221.
Facchetti F., Blanzuoli L., Ungari M., Alebardi O., and Vermi W. 1998. Lymph node pathology in primary combined immunodeficiency diseases. Springer Semin. Immunopathol. 19(4): 459–478.
Falini B., Fizzotti M., Pucciarini A., Bigerna B., Marafioti T., Gambacorta M., Pacini R., Alunni C., Natali-Tanci L., Ugolini B., Sebastiani C., Cattoretti G., Pileri S., Dalla-Favera R., and Stein H. 2000. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood, 95(6): 2084–2092.
Gilmore T.D. 2006. Introduction to NF-κB: Players, pathways, perspectives. Oncogene, 25(51): 6680–6684.
Gualco G., Weiss L.M., and Bacchi C.E. 2010. MUM1/IRF4: A review. Appl. Immunohistochem. Mol. Morphol. 18(4): 301–310.
Kaustio M., Haapaniemi E., Göös H., Hautala T., Park G., Syrjänen J., Einarsdottir E., Sahu B., Kilpinen S., Rounioja S., Fogarty C.L., Glumoff V., Kulmala P., Katayama S., Tamene F., Trotta L., Morgunova E., Krjutškov K., Nurmi K., Eklund K., Lagerstedt A., Helminen M., Martelius T., Mustjoki S., Taipale J., Saarela J., Kere J., Varjosalo M., and Seppänen M. 2017. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J. Allergy Clin. Immunol. 140(3): 782–796.
Lo J.C., Basak S., James E.S., Quiambo R.S., Kinsella M.C., Alegre M.L., Weih F., Franzoso G., Hoffmann A., and Fu Y.X. 2006. Coordination between NF-κB family members p50 and p52 is essential for mediating LTβR signals in the development and organization of secondary lymphoid tissues. Blood, 107(3): 1048–1055.
Lorenzini T., Fliegauf M., Klammer N., Frede N., Proietti M., Bulashevska A., Camacho-Ordonez N., Varjosalo M., Kinnunen M., de Vries E., van der Meer J.W.M., Ameratunga R., Roifman C.M., Schejter Y.D., Kobbe R., Hautala T., Atschekzei F., Schmidt R.E., Schröder C., Stepensky P., Shadur B., Pedroza L.A., van der Flier M., Martínez-Gallo M., Gonzalez-Granado L.I., Allende L.M., Shcherbina A., Kuzmenko N., Zakharova V., Neves J.F., Svec P., Fischer U., Ip W., Bartsch O., Barış S., Klein C., Geha R., Chou J., Alosaimi M., Weintraub L., Boztug K., Hirschmugl T., Dos Santos Vilela M.M., Holzinger D., Seidl M., Lougaris V., Plebani A., Alsina L., Piquer-Gibert M., Deyà-Martínez A., Slade C.A., Aghamohammadi A., Abolhassani H., Hammarström L., Kuismin O., Helminen M., Allen H.L., Thaventhiran J.E., Freeman A.F., Cook M., Bakhtiar S., Christiansen M., Cunningham-Rundles C., Patel N.C., Rae W., Niehues T., Brauer N., Syrjänen J., Seppänen M.R.J., Burns S.O., Tuijnenburg P., and Kuijpers T.W.NIHR BioResource,Warnatz K. and Grimbacher B. 2020. Characterization of the clinical and immunologic phenotype and management of 157 individuals with 56 distinct heterozygous NFKB1 mutations. J. Allergy Clin. Immunol. 146(4): 901–911.
Mandola A.B., Sharfe N., Nagdi Z., Dadi H., Vong L., Merico D., Ngan B., Reid B., and Roifman C.M. 2021. Combined immunodeficiency caused by a novel homozygous NFKB1 mutation. J. Allergy Clin. Immunol. 147(2): 727–733.e2.
Ratech H., Hirschhorn R., and Greco M.A. 1989. Pathologic findings in adenosine deaminase deficient-severe combined immunodeficiency. II. Thymus, spleen, lymph node, and gastrointestinal tract lymphoid tissue alterations. Am. J. Pathol. 135(6): 1145–1156.
Smart B.A. 2014. Deficiency of innate and acquired immunity caused by an IKBKB mutation. Pediatrics, 134(Suppl. 3): S181.
Tuijnenburg P., Lango Allen H., Burns S.O., Greene D., Jansen M.H., Staples E., Stephens J., Carss K.J., Biasci D., Baxendale H., Thomas M., Chandra A., Kiani-Alikhan S., Longhurst H.J., Seneviratne S.L., Oksenhendler E., Simeoni I., de Bree G.J., Tool A.T.J., van Leeuwen E.M.M., Ebberink E.H.T.M., Meijer A.B., Tuna S., Whitehorn D., Brown M., Turro E., Thrasher A.J., Smith K.G.C., Thaventhiran J.E., and Kuijpers T.W.NIHR BioResource–Rare Diseases Consortium. 2018. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J. Allergy Clin. Immunol. 142(1): 1285–1296.
Weih D.S., Yilmaz Z.B., and Weih F. 2001. Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J. Immunol. 167(4): 1909–1919.
Willard-Mack C.L. 2006. Normal structure, function, and histology of lymph nodes. Toxicol. Pathol. 34(5): 409–424.
Zhang Q., Lenardo M.J., and Baltimore D. 2017. 30 years of NF-κB: A blossoming of relevance to human pathobiology. Cell, 168(1–2): 37–57.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 8 • Number 1 • March 2021
Pages: 11 - 18
History
Received: 28 January 2021
Accepted: 23 February 2021
Accepted manuscript online: 24 February 2021
Copyright
© 2021.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
JennyGarkaby and BoNgan. 2021. Homozygous NF-kB1 mutation causing combined immunodeficiency: a histopathological analysis. LymphoSign Journal.
8(1): 11-18. https://doi.org/10.14785/lymphosign-2021-0012
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
