Radiographic features of patients with chronic granulomatous disease
Abstract
Introduction: Chronic granulomatous disease (CGD) is one of the most common primary immunodeficiencies of childhood, and is caused by defects in the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. Alongside neutrophil dysfunction, dysregulation of the immune system predisposes patients to recurrent life-threatening infections as well as granuloma formation, hyperinflammation, and autoimmunity. Examination by imaging (radiography, ultrasound, computed tomography, magnetic resonance) in conjunction with biopsy and tissue or fluid cultures are essential to identify the extent and severity of infections as well as the microorganisms responsible. These modalities also help to guide the management of inflammatory complications.
Aim: We highlight the common radiographic findings in 10 pediatric CGD patients followed at our centre over a period of 10 years.
Methods: Medical records of patients with confirmed CGD diagnosis were reviewed retrospectively. All had low neutrophil oxidative burst index (NOBI) and pathogenic mutation in 1 of the 5 subunits of the NADPH oxidase. Three patients had autosomal recessive CGD and 7 had X-linked recessive CGD. All but 1 are male.
Results: The most common radiographic presentation was hilar lymphadenopathy and pulmonary nodules. Other lung complications include cavitating lesions, lung abscess, pulmonary nodule, and pleuritic nodules. Lymphatic tissue and lymph nodes were involved in 50% of our cohort of patients, while gastrointestinal manifestations were noted in approximately 35% of our patients. These include the presence of pigmented macrophages, multiple granulomas, liver abscess, or detection of Aspergillus in tissue or fluid culture.
Discussion: It is essential for clinicians to keep primary immunodeficiency as one of the differential diagnoses in patients who present with severe infection or inflammation. We encourage physicians to consider CGD in patients with above described findings and consider measuring NOBI in patients with early onset infection, inflammation, or granuloma formation.
Statement of novelty: We describe the radiographic findings of a pediatric cohort of patients with CGD.
Introduction
Chronic granulomatous disease (CGD) is one of the most common primary immunodeficiencies of childhood, often diagnosed within the first 3 years of life (Kuhns et al. 2010; Battersby et al. 2013; Thomsen et al. 2016). It is caused by defects in the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, required for phagocytic production of reactive oxygen species (ROS) and subsequent intracellular killing of bacteria and fungi. Any mutation within the genes encoding the 5 subunits of the NADPH oxidase, namely CYBB (gp91phox), NCF1 (p47phox), NCF2 (p67phox), NCF4 (p40phox), and CYBA (p22phox), resulting in functional impairment of ROS production, can lead to CGD (Matute et al. 2009; Roos and de Boer 2014).
Patients commonly present with lower respiratory tract infections, acute or recurrent bacterial and fungal infections, or failure to thrive (Guide et al. 2003; Thomsen et al. 2016). Due to both neutrophil dysfunction and dysregulation of the immune system, affected individuals are predisposed to granuloma formation, hyperinflammation, and autoimmunity (Cale et al. 2000; Winkelstein et al. 2000; De Ravin et al. 2008). Manifestations in the gastrointestinal tract (early onset inflammatory bowel disease) can be another indication of CGD (Marciano et al. 2004). Although symptoms often appear during infancy, in some, presentation occurs during late childhood or adulthood and is associated with residual function of the NADPH oxidase system (Köker et al. 2013). Patients with CGD may also be affected by other auto-inflammatory disorders, including systemic lupus erythematous, arthritis, or vasculitis. Osteomyelitis is occasionally seen as well. Dysfunctional autophagy and inflammasome activation as well as inadequate resolution of inflammation are thought to contribute to the pathophysiology of CGD.
Defects in NADPH oxidase confer infectious susceptibility to a surprisingly limited spectrum of microbes; specifically, those that are catalase-positive (Kaplan et al. 1968; Lazarus and Neu 1975). Catalase degrades host-derived hydrogen peroxide, thereby limiting the availability of ROS to kill phagocytosed organisms. For this reason, infections of the lung associated with Aspergillus fumigatus account for up to 40% of cases, while infections stemming from Staphylococcus aureus and Burkholderia cepacia are also common. Liver abscesses occur in almost 35% of cases, with skin and lymph node infection taking place in approximately 20% of patients. In North America, the majority of serious infections are caused by Staphylococcus aureus, Serratia marcescens, Burkholderia cepacia complex, Aspergillus species, Nocardia species, and Klebsiella species, although involvement of other microorganisms have also been reported.
Importantly, inflammatory manifestations in the absence of underlying infection can affect a variety of organs and tissues, including the brain, lungs, liver, spleen, gastrointestinal tract, genitourinary tract, skin, and lymph nodes (Marciano et al. 2004; Mahdaviani et al. 2013). Non-caseating granulomatous lesions composed of multinucleated giant cells are a classic finding in patients with CGD (Schappi et al. 2008). In the lungs, liver, and spleen, such lesions may flare and resolve spontaneously, without intervention. Thus, examination by imaging (radiography, ultrasound, computed tomography, magnetic resonance), alongside biopsy and tissue or fluid cultures are essential to identify the extent and severity of infections as well as the microorganism responsible. These modalities also help to guide the management of inflammatory complications.
Once clinical suspicion of CGD arises, the diagnosis is investigated by assessing the functional activity of NADPH oxidase. This can be achieved by direct measurement of superoxide (for example, nitroblue tetrazolium (NBT) reduction) or by flow cytometric analysis of dihydrorhodamine (DHR) oxidation, which measures neutrophil oxidative burst function. The latter method provides a ratio (termed neutrophil oxidative burst index, NOBI) of oxidative function in neutrophils of the suspected case versus healthy control.
Genetic diagnosis of CGD is considered standard of care and is essential for prognosis, long term care planning, and family counselling. Mutations in CYBB, located on the X chromosome and thus conferring X-linked recessive inheritance, account for up to 65% of CGD cases in North America. In contrast, an estimated 25% of cases stem from biallelic mutations in NCF1, which is inherited in an autosomal recessive manner. Mutations in CYBA, NCF2, and NCF4 represent <5% of CGD cases. X-linked CGD is considered more clinically severe, with earlier presentation and more severe infections than autosomal recessive CGD (Winkelstein et al. 2000; Kuhns et al. 2010).
Female carriers of a single CYBB mutation have impaired NADPH oxidase activity in only a portion of phagocytes as a result of lyonization. This active carrier state can lead to a range of symptoms, including aphthous ulcers, arthralgia, and cutaneous photosensitivity (Battersby et al. 2013). Infectious complications are known to be highly likely in patients with low residual neutrophil oxidative burst function (Kuhns et al. 2010; Marciano et al. 2018).
Prophylactic treatment with trimethoprim-sulfamethoxazole and an azole antifungal agent, such as itraconazole, is the most effective modality of treatment to reduce the frequency of potentially fatal bacterial and fungal infections. It is important to note that despite appropriate prophylaxis, many patients still experience at least 1 severe bacterial or fungal infection every 3–4 years (Roos and de Boer 2014).
Currently, allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative option for patients with CGD (Ho et al. 1996; Seger et al. 2002; Ahlin et al. 2013). The HSCT event-free survival rate for CGD patients is estimated >80%, with outcomes likely to continue to improve over time (Connelly et al. 2018). In contrast, results of gene therapy trials have so far been discouraging due to associated myelodysplastic syndrome, difficulty achieving long-term engraftment, and complications such as oligoclonal expansion (Kohn 2010; Kaufmann et al. 2014).
Here, we present radiographic findings of 10 pediatric patients who were diagnosed with CGD. These patients were followed in our centre over a period of 10 years. Seven had an X-linked form of CGD with confirmed mutations in CYBB, the gene encoding gp91phox. Three patients had autosomal recessive CGD; 2 of them with mutations in NCF1 encoding subunit p47phox of NADPH oxidase, and 1 with a rare mutation in NCF2, which encodes p67phox. Patient characteristics are summarized in Table 1.
Table 1:
Note: NOBI, neutrophil oxidative burst index; NA, not available.
Case presentation
Patient 1
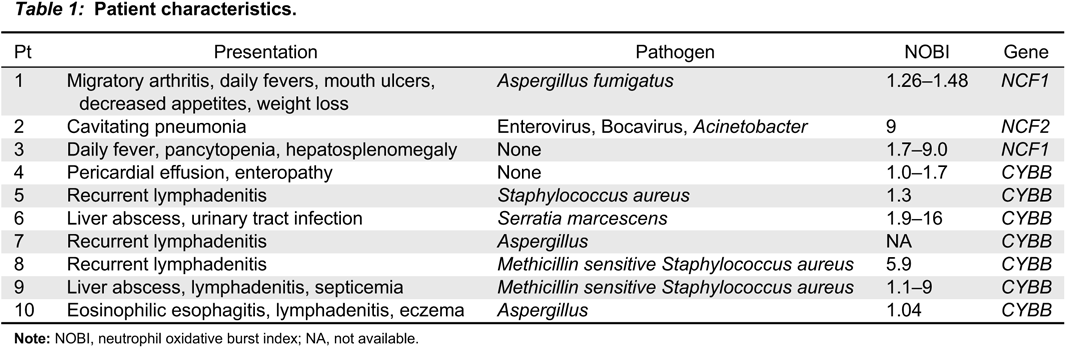
Patient 1, a previously healthy male, presented at 12 years of age with migratory arthritis, swelling of fingers and toes, mouth ulcers, decreased appetite, weight loss, and daily fevers. He was admitted to hospital where initial investigations showed signs of macrophage activation syndrome. He was treated with high dose prednisone, and his symptoms progressively resolved. Two months later, he was re-admitted with fevers, painful oral ulcers, cough, and pleuritic chest pain. Computed tomography (CT) scan of his chest showed bilateral pulmonary nodules with mediastinal and hilar lymphadenopathy (Figure 1). A bronchoalveolar lavage was positive for Aspergillus fumigatus. He was diagnosed with autosomal recessive CGD after genetic assessment revealed a mutation in NCF1.
Figure 1:

Patient 2
Patient 2 presented at 1 year of age with prolonged fever and poor response to 1 course of antibiotics. Further evaluation revealed a right lower lobe cavitating pneumonia and hilar mediastinal lymphadenopathy, found to be positive for Enterovirus, Bocavirus, and Acinetobacter. His tuberculosis (TB) skin test was negative and gastric aspirate was twice negative. Follow up chest CT demonstrated worsening of the right lower lobe opacity with small confluent cavitation and associated hilar and mediastinal lymphadenopathy (Figure 2). He was treated with multiple courses of antimicrobials including meropenem. Genetic evaluation revealed a mutation in NCF2, confirming the diagnosis of autosomal recessive CGD. He later developed recurrent mouth ulcers on the inside of his lower lip, which resolved over 4–5 days without treatment. This remains an active issue for him.
Figure 2:
Patient 3
Patient 3 is the only female patient in our case series. At 2 years of age, she had a history of afebrile cervical adenitis with spontaneous non-purulent drainage that resolved with a course of amoxicillin-clavulanate. Aside from 3 episodes of acute otitis media responsive to short course of antibiotics, there was no other significant infection history. She had oral aphthous lesions in childhood that resolved over years, as well as recurrent, prolonged fevers ranging from 6 days to 2 weeks. At 7 years of age, she presented with decreased appetite, abdominal pain and fullness, as well as generalized fatigue. These episodes were found to be associated with significant splenomegaly and moderate hepatomegaly as well as pan or bi-cytopenia. Infectious work up was persistently negative. Bone marrow biopsy showed mild fibrosis with no evidence of malignancy or hemophagocytosis. Her full body magnetic resonance imaging (MRI) and abdominal ultrasound revealed hepatosplenomegaly but no other abnormal finding. Splenic biopsy identified diffuse infiltrate with small, well-defined non-caseating granulomas. The patient was diagnosed with juvenile sarcoidosis, and ophthalmology assessment showed uveitis. She was subsequently started on oral prednisone (2 mg/kg), ocular prednisone, and methotrexate. This resulted in marked improvement, with resolution of cytopenia, fevers, hepatosplenomegaly, and uveitis. Given the presence of granulomas in the spleen and low NOBI (from 1.7 to 9.0) on 3 separate occasions, genetic testing was subsequently arranged. A mutation in NCF1 confirmed the diagnosis of autosomal recessive CGD. Antimicrobial and antifungal prophylaxis was initiated, however, with poor compliance. Upon cessation of immunosuppression, she had resurgence of mild cytopenia and hepatosplenomegaly. She had another flare of fever, splenomegaly, and abdominal pain a year later associated with severe headache and papilledema. Brain MRI and subsequent magnetic resonance venography (MRV) showed evidence of increased intracranial pressure, and abdominal MRI (Figure 3) showed an enlarged liver and spleen with with parenchymal heterogeneity in the periphery of the spleen, as well as some prominent periportal and peripancreatic enlarged lymph nodes. Lumbar puncture showed increased opening pressure (37) with benign cerebrospinal fluid (CSF) examination. She was started on acetazolamide with somewhat adequate response, and receives low dose steroids in case of flare-ups. The patient has not been transplanted, however, this is considered an option for her.
Figure 3:

Patient 4
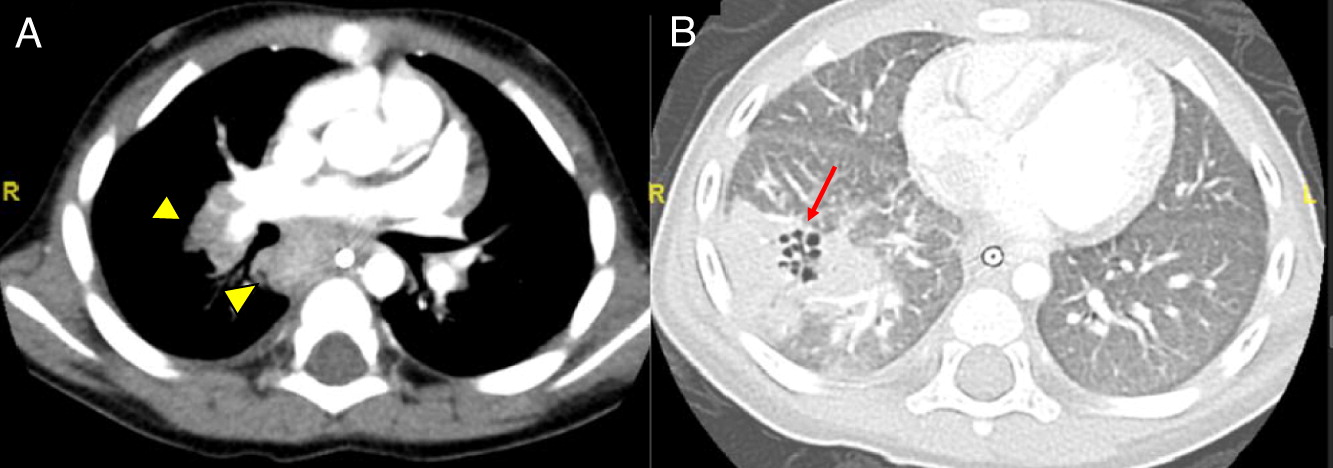
Patient 4 developed idiopathic serous pericardial effusion with tamponade at age 19 months, requiring pericardiocentesis. At 3 years of age, he had significant episodes of vomiting, abdominal pain, and severe obstruction. Ultrasound and CT scan of the abdomen was suggestive of multiple sites of inflammation from the esophagus including the small and large bowel (Figure 4). Biopsies revealed focal eosinophilic abscess in the duodenum but no granulomas, as well as multiple pigmented macrophages in the lamina propria. He was started on omeprazole and ketotifen for eosinophilic gastroenteritis, but his weight loss and vomiting persisted. Repeat upper and lower gastrointestinal (GI) endoscopies and biopsies of the colon and rectum revealed positive Periodic acid-Schiff (PAS) staining of macrophages in the rectum, raising the possibility of CGD. Vomiting was explained by gastric outlet obstruction, a gastrojejunal tube was inserted for feeding, and he was started on steroids for the GI inflammation. The patient had abnormal NOBI on 2 occasions at levels of 1.0 and 1.7, while the patient’s mom was found to have 2 populations of neutrophils with normal and abnormal NOBI. Genetic testing was confirmatory of X-linked CGD with a point mutation in CYBB. He was subsequently started on antimicrobial and antifungal prophylaxis. The patient had brain MRI and angiography due to refractory and persistent vomiting which showed irregular asymmetric dilatation of basilar artery (Figure 5). No intervention was necessary. He had flare ups of abdominal pain 4–5 times a year which responded well to steroids. Although he had a human leukocyte antigen (HLA)-matched sibling, the family did not consent to HSCT. He was lost to follow-up for several years, and was admitted at the age of 12 years to the intensive care unit with septicemia secondary to small bowel microperforations requiring small bowel resection and ileostomy. The resected portion of small bowel, as well as biopsies from other GI sites, showed non-necrotizing granulomas. The procedure was later complicated with an abdominal abscess. Subsequent MRI of the pelvis revealed persistent bowel inflammation mimicking IBD. A CT scan of his chest showed multiple nodules/granulomas in his right lung. The patient later died due to sepsis and respiratory failure.
Figure 4:
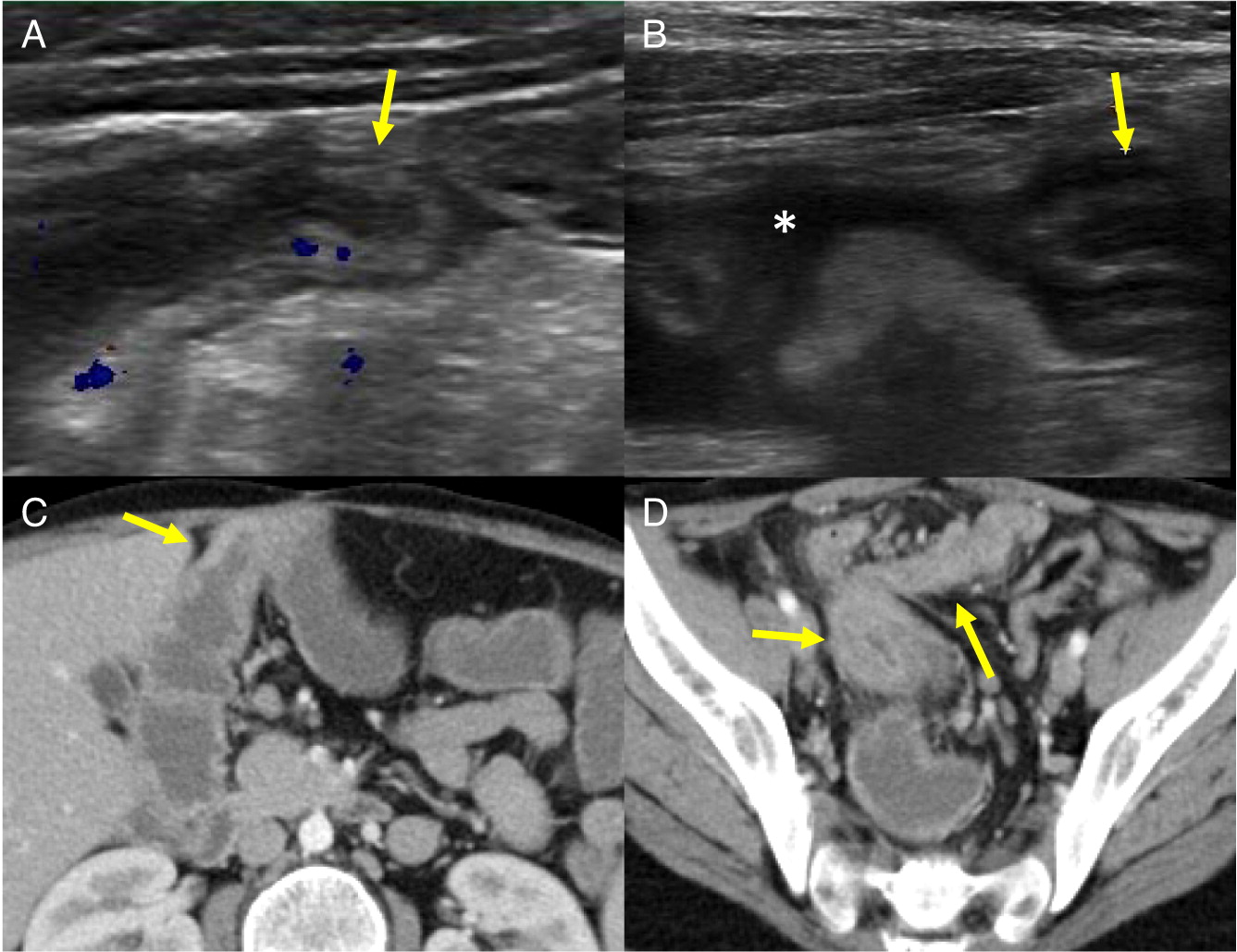
Figure 5:

Patient 5
Patient 5 presented at 2 months of age with cervical lymphadenopathy on the left side requiring excision and debridement due to abscess formation. Cultures grew Staphylococcus aureus and the tissue showed granuloma formation. He presented again at 4.5 months of age with bilateral neck lymphadenopathy (Figure 6) and fever. Cultures again grew S. aureus and the microscopic description was consistent with granulomatous inflammation. NOBI was low at 1.3, and alongside the presence of granuloma and frequent lymphadenitis, this lead to the diagnosis of X-linked CGD. A novel mutation in CYBB encoding for gp91phox was identified. Given the persistence of fever, a CT of his chest was performed which showed multiple small pulmonary nodules (Figure 7) and an abdominal ultrasound was also performed which demonstrate multiple hypoechoic splenic lesions in a mildly enlarged spleen (Figure 8). Once his lymphadenitis and fever improved, he was started on trimethoprim-sulfamethoxazole and itraconazole for bacterial and fungal prophylaxis. He underwent a successful matched sibling donor HSCT at 9 months of age and has full immune reconstitution.
Figure 6:
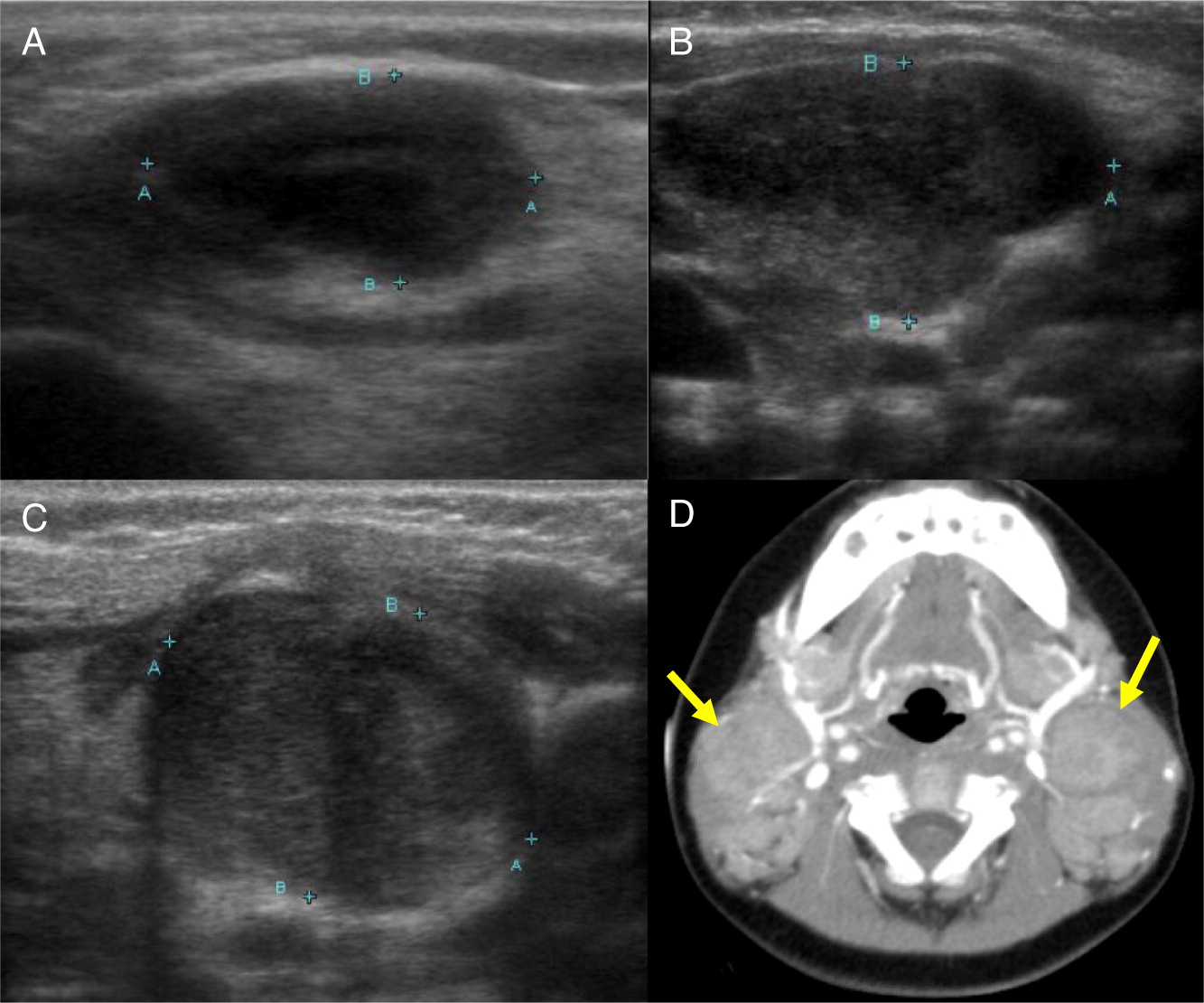
Figure 7:
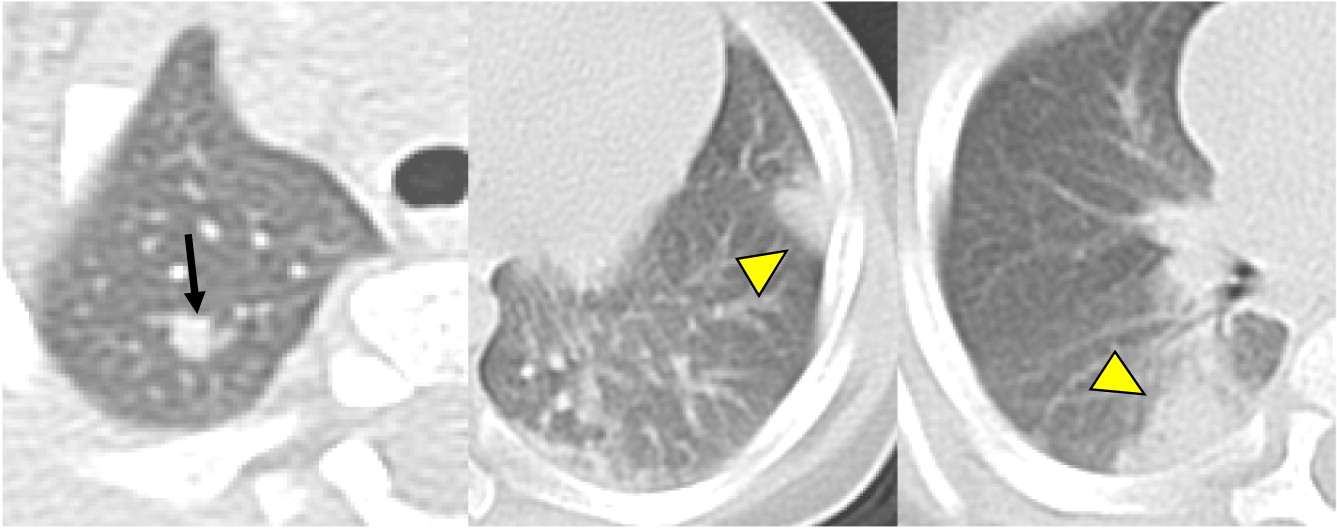
Figure 8:

Patient 6
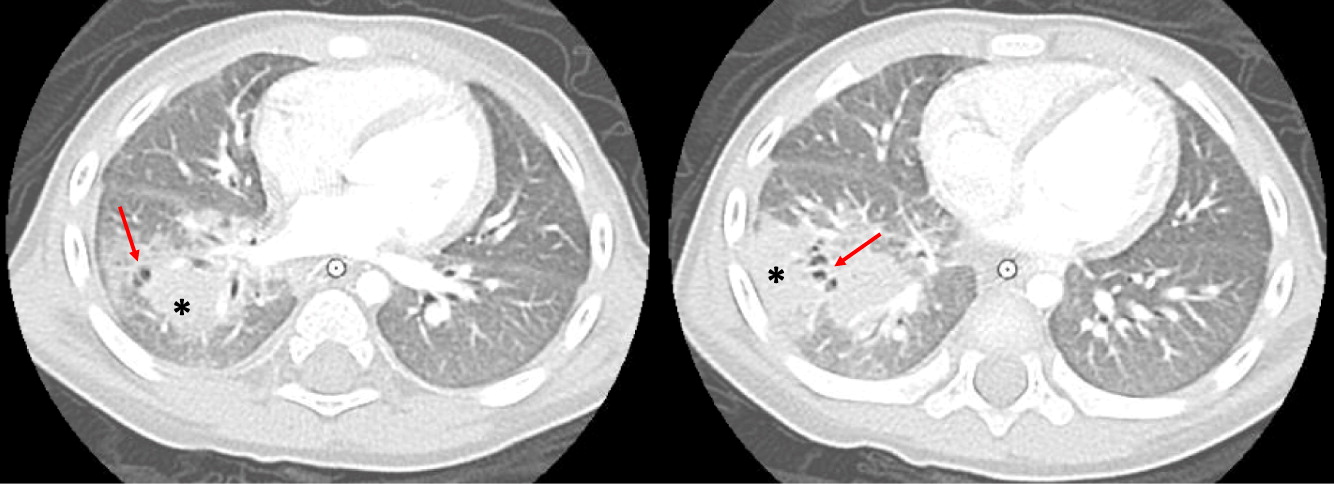
Patient 6 presented at 18 months of age with a history of diarrhea and fever. An abdominal ultrasound and CT scan were performed, showing 2 hypoechoic lesions in his liver (Figures 9A and 9B). He was treated with cefotaxime, ampicillin, and flagyl, then subsequently switched to cloxacillin and ciprofloxacin. Biopsy of the lesions showed non-necrotizing granulomatous inflammation. He continued to have persistent fevers and was eventually found to have Serratia marcescens in a urine culture. Further imaging with an abdominal CT showed diffuse large bowel wall thickening (Figure 9C). An upper and lower endoscopy revealed severe colitis, particularly in the proximal bowel. He was given mesalamine as well as interferon gamma 3 times a week while he was admitted in hospital. A CT of his chest showed scattered small lesions in the posterior area of both lungs (Figure 10). He had 2 low NOBI and subsequent genetic testing confirmed X-linked CGD due to mutation in the CYBB gene. His mother was identified as a carrier. Once the infections were controlled, he underwent a successful matched sibling donor HSCT. Unfortunately, he had multiple issues post-transplant, including hypothyroidism, short stature, bronchiolitis obliterans requiring steroid treatment, ADHD, learning disability, recurrence of his colitis, bilateral lichen sclerosis atrophicus of axilla, and plasmacytoma of his tongue. He also had disseminated measles after MMR vaccination that required hospital admission. The patient remains partially engrafted.
Figure 9:
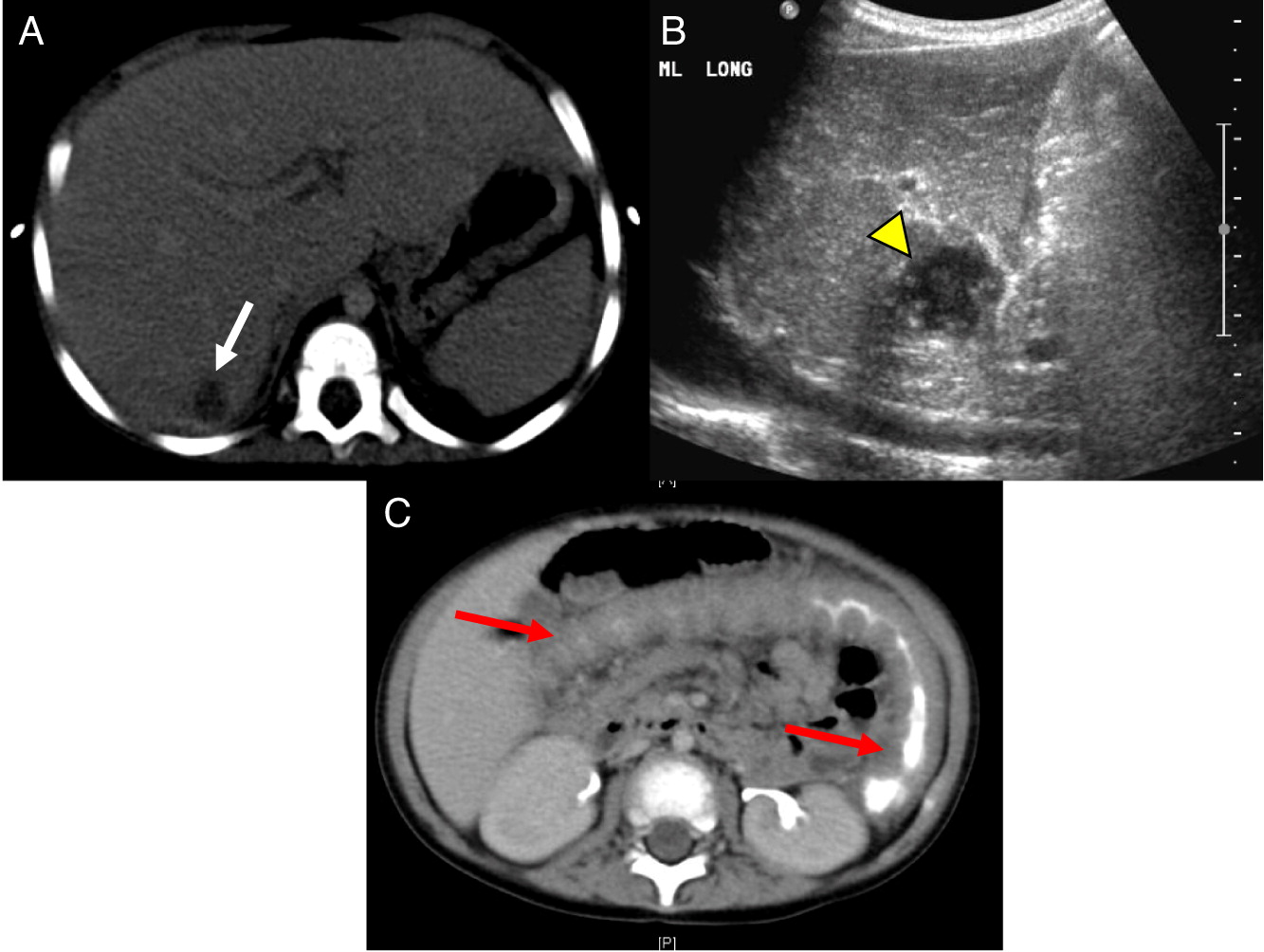
Figure 10:
Patient 7
Patient 7 presented at 1 year of age with cervical lymphadenitis requiring excision. At 3 years of age he developed multiple left axillary enlarged lymph nodes with surrounding inflammatory changes (Figure 11) and an erythematous nodule on his left arm. This lymph node was excised and grew Aspergillus. This finding raised the suspicion of CGD. Further laboratory testing and genetic work up confirmed the diagnosis of X-linked CGD due to mutation in CYBB. A CT scan of his chest demonstrated mediastinal and hilar lymphadenopathy as well as extensive nodular lung disease (Figure 12). He was treated with amphotericin and later with voriconazole and itraconazole, with excellent response to therapy after 4 months. Repeat imaging showed complete resolution of the axillary lesion and a significant decrease in the number of lung lesions. Six months later, abdominal ultrasound showed multiple splenic lesions and splenomegaly (Figure 13). He was treated again with amphotericin and caspofungin, however, the lesions did not improve and continued to wax and wane over the following months. A laparoscopic biopsy was then performed, which was negative for fungi or bacteria but showed non-necrotizing granulomas. He was started on weekly interferon gamma therapy and eventually received a matched sibling donor HSCT. He was fully engrafted and all lesions completely resolved. Unfortunately, he developed rhabdomyosarcoma of the left posterior fossa which was treated with surgery, chemotherapy and radiation. Subsequently, he developed hypothyroidism and is being treated with thyroid hormone replacement therapy.
Figure 11:
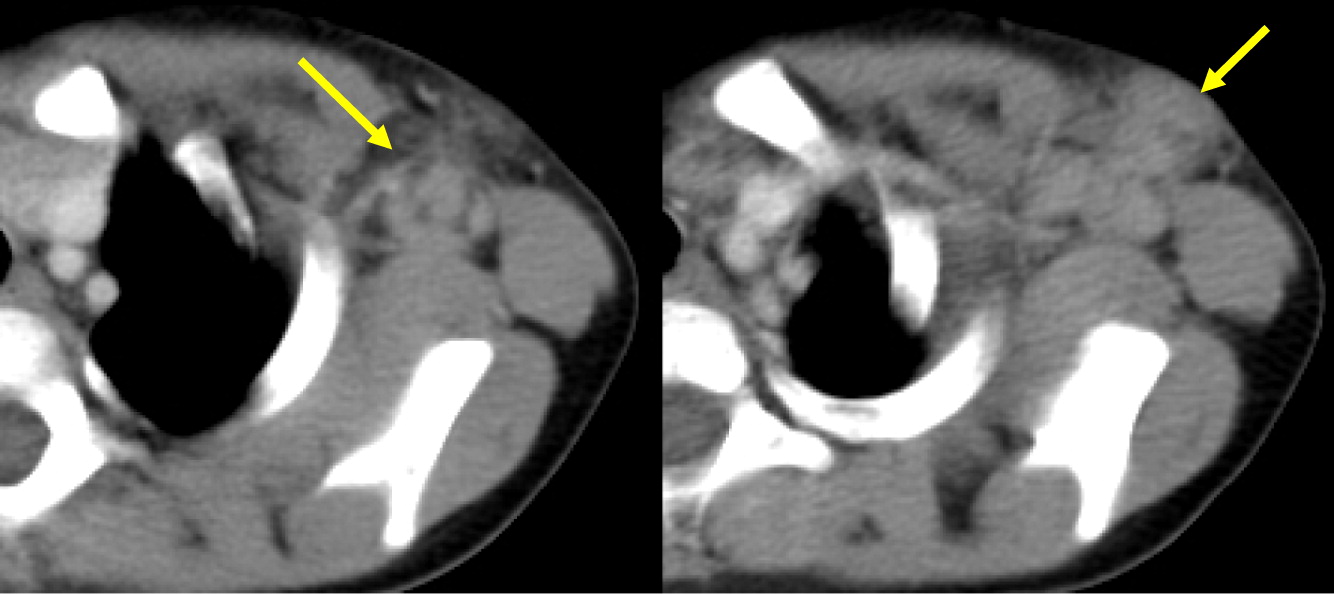
Figure 12:

Figure 13:
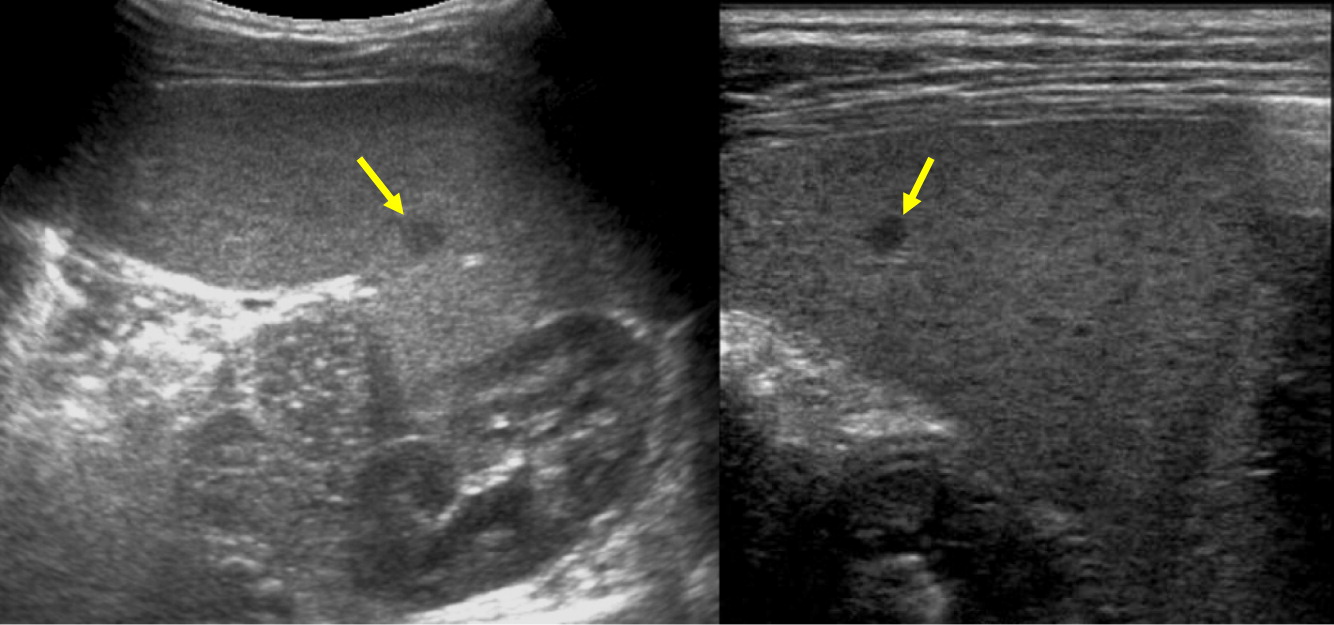
Patient 8
Patient 8 presented at 6 months of age with severe skin impetigo that was treated with antibiotics. He has had 2 episodes of left sided cervical lymphadenitis (Figure 14). The first was at 8 months of age and required operative management. Cultures grew methicillin sensitive Staphylococcus aureus (MSSA). The second lymphadenitis was at 19 months old, however, no pathogen was identified. His most recent NOBI result was 5.9. Genetic testing confirmed X-linked CGD due to mutation in CYBB. He later underwent successful HSCT from a 9/10 HLA-matched unrelated donor.
Figure 14:
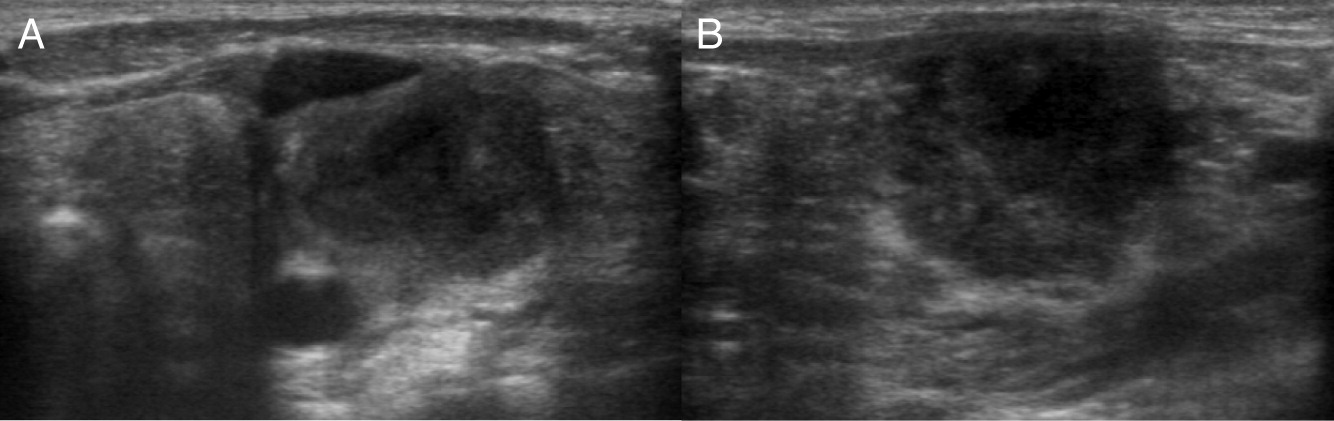
Patient 9
Patient 9 presented at 3 months of age with fever, poor weight gain, and irritability. Hospital admission and investigations revealed septicemia (blood culture grew MSSA), bilateral neck lymphadenitis (Figure 15) and liver abscess (Figure 16). His liver abscess was biopsied and showed granuloma inflammation as well as positive culture for MSSA. For this, he was treated with a prolonged course of antibiotics. The patient’s NOBI test result was abnormally low and subsequent genetic testing identified a mutation in CYBB, confirming X-linked CGD. He received HSCT from 9/10 HLA-matched unrelated donor. His post-transplant course was complicated with liver and GI graft versus host disease (GvHD), requiring multiple immunosuppressive treatments to control his symptoms. To date, the patient’s GI GvHD has not flared for over a year, however, he is still receiving immunosuppression for his liver GvHD.
Figure 15:
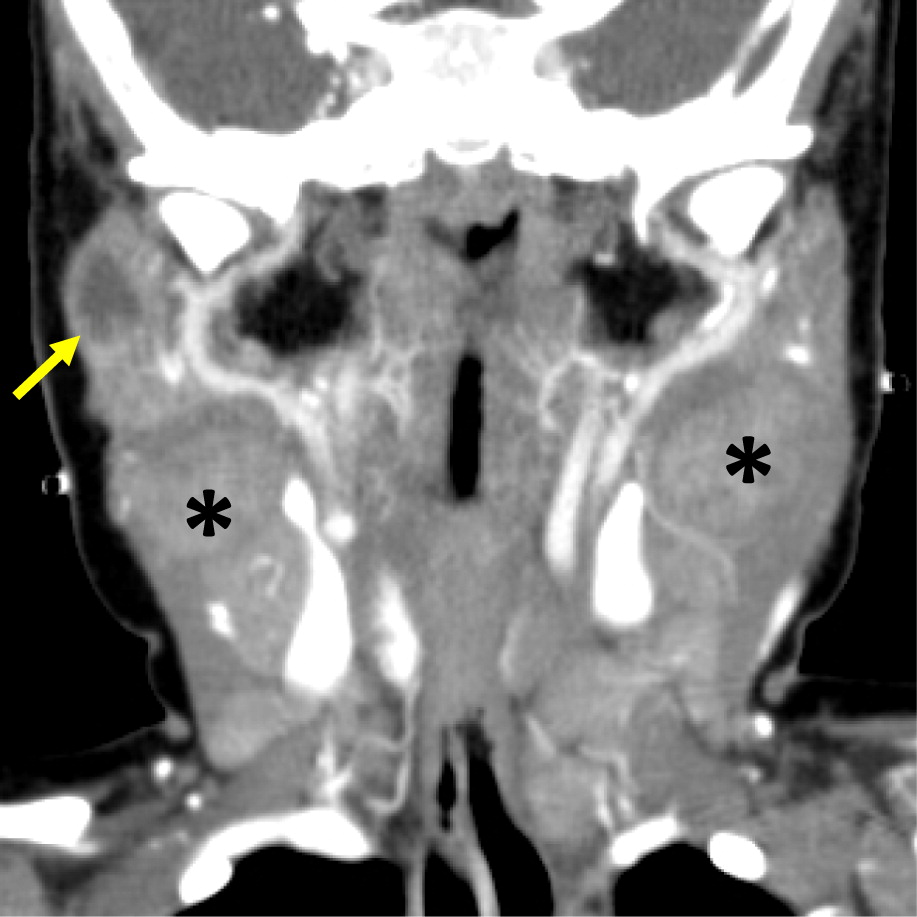
Figure 16:
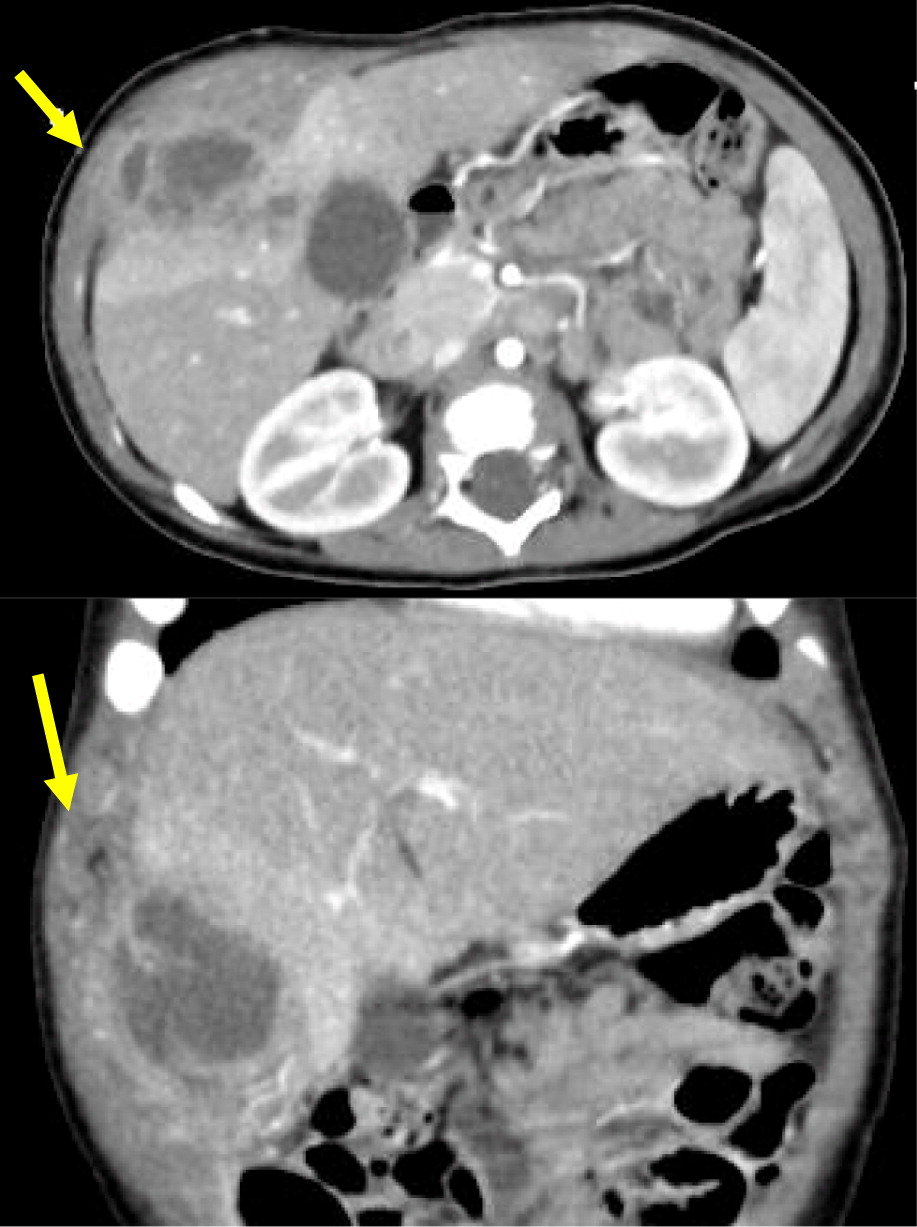
Patient 10
Patient 10 presented at 3 days of age with diarrhea and severe eczema, and was diagnosed with eosinophilic gastroenteritis. At 10 months of age, he developed bilateral cervical lymphadenitis. Lymph node excision showed granulomatous inflammation with no organism identified. Subsequent genetic testing revealed mutation in CYBB, confirming the diagnosis of X-linked CGD. The patient was started on prophylactic antibiotic, antifungals, and interferon gamma, however, he experienced recurrent mouth ulcers and intermittent anal fissures, as well as recurrent skin infections and abscesses. The patient decided to discontinue interferon gamma at 11 years of age, and prophylactic antibiotic and antifungals at 14 years of age. At 15, he was admitted with an abscess of his right foot, right thigh, and right lung. A CT scan of his lungs revealed consolidation of the right middle and lower lobes with mild pleural effusion, as well as mediastinal and hilar lymphadenopathy (Figure 17). Follow up chest MRI showed progression of the lung consolidation to an intrapulmonary abscess extending to the thoracic wall (Figure 18). A lung biopsy revealed Aspergillus. MRI of lower extremity showed an inflammatory/infective process in his right thigh (Figure 19). Skin biopsies from his right foot and from his right thigh revealed abscesses with granulomatous inflammation without any microorganism identified.
Figure 17:

Figure 18:
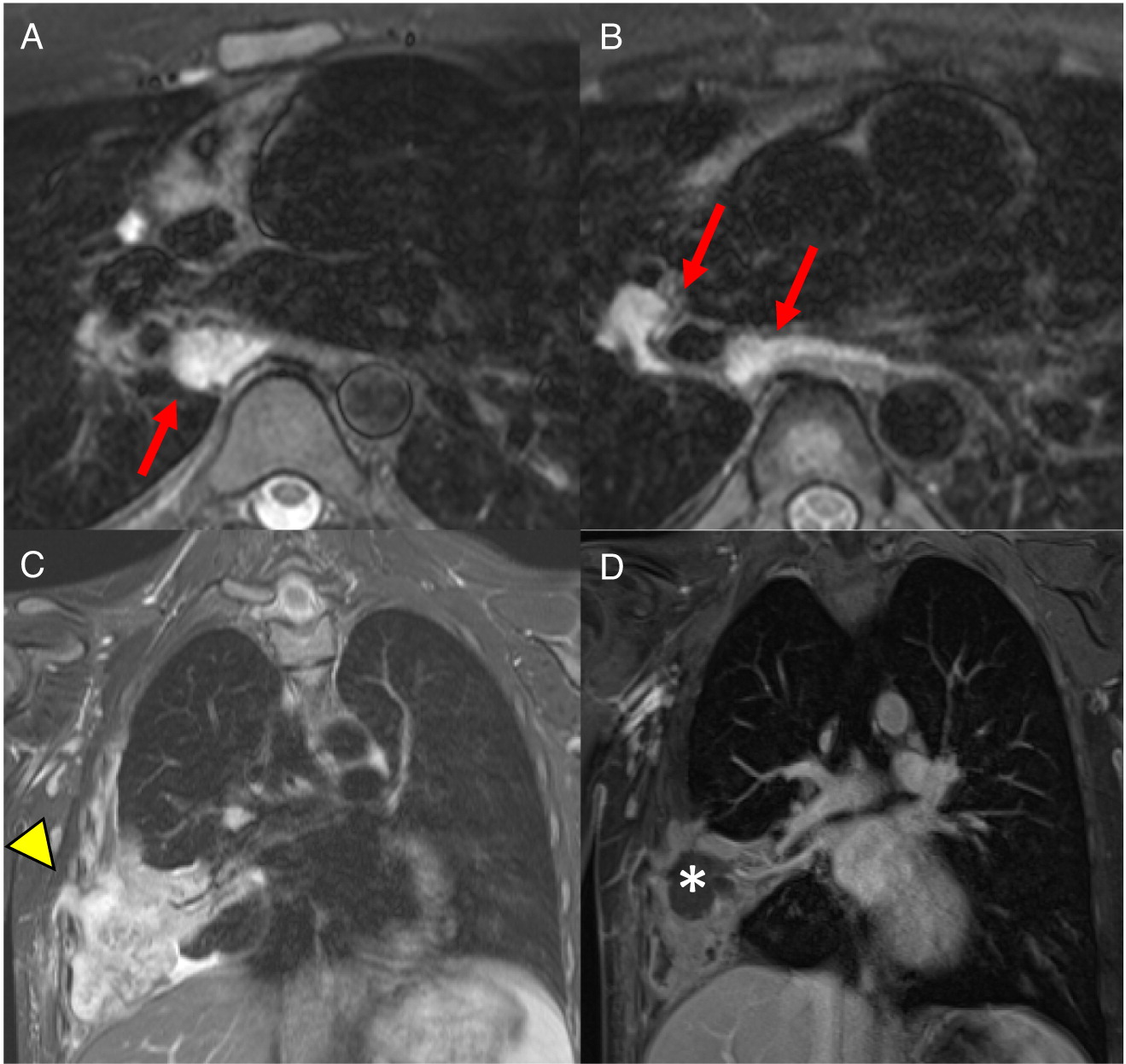
Figure 19:

Discussion
As illustrated in this case series, invasive bacterial and fungal infections as well as inflammatory complications are a significant threat for CGD patients. Radiological assessment of patients who present with recurrent infection is extremely helpful for guiding diagnosis and monitoring the response for treatment (Towbin and Chaves 2010). Very often, these cases require biopsy or culture to isolate the causative microorganism. Together, this can further guide antimicrobial and antifungal therapy, and help clinicians to better understand the underlying immunodeficiency.
Imaging of the lungs is one of the most important modalities to consider. In this case series, 8 of 10 patients had pulmonary manifestations such as pulmonary nodule, abscess, cavitation or hilar lymphadenopathy. A combination of these findings in any 1 patient is suggestive of underlying immunodeficiency. As many as 80% of CGD patients present with pulmonary complications and this remains a primary cause of death in non-transplanted patients (Winkelstein et al. 2000). Chest radiographs may show a variety of findings including consolidations, reticulonodular opacities, bronchiectasis, and scarring. CT scans may also identify consolidation, ground-glass opacities, centrilobular nodules, bronchiectasis, septal thickening, air trapping, or scarring. Complications such as abscess formation, cavitary lesions, empyema, or lung fibrosis may also be shown. There have also been reports of spreading to the chest wall and associated osteomyelitis of ribs and vertebrae (Towbin and Chaves 2010; Mahdaviani et al. 2013).
Changes in lymph nodes, particularly lymphadenopathy, lymphadenitis, and granuloma formation was the second most common finding in the CGD patients reviewed here. The cervical chain is most frequently involved, alongside hilar and mediastinal lymph nodes.
Hepatic abscesses or granulomas are often found in the liver, and are present in 25%–50% patients. Although liver lesions are more common in the adult population, they can found in pediatric patients as well. In our patients, 2 had liver abscesses and (or) granuloma. As demonstrated in our cohort, such lesions may also affect the spleen, and tend to wax and wane over time, generally disappearing or shrinking after HSCT (Winkelstein et al. 2000; Towbin and Chaves 2010).
The most common imaging findings of the GI tract are bowel wall thickening and inflammation mimicking IBD. In our patients, 3 out of 10 had GI manifestations identified upon biopsy or bowel imaging. These vary from granuloma formation to non-specific bowel wall thickening or inflammation. The age of presentation (early onset IBD) and presence of pigmented macrophages are 2 important clues for diagnosis of CGD. Interestingly, the muscle lesions seen in one of our patients is not frequently reported (Murguia-Favela and Manson 2014).
To date, advancements in antimicrobial and antifungal prophylaxis have been essential for improving the morbidity and mortality of patients with CGD. Radiographic and pathological findings continue to be essential for monitoring disease progression, while genetic testing of the patient and family members can aid in the determination of carrier status. Furthermore, tests that evaluate the level of residual NADPH function and thus severity of disease can be used to identify patients who would benefit from HSCT.
REFERENCES
Ahlin A., Fugelang J., de Boer M., Ringden O., Fasth A., and Winiarski J. 2013. Chronic granulomatous disease-haematopoietic stem cell transplantation versus conventional treatment. Acta Paediatr. 102: 1087–1094.
Battersby A.C., Cale A.M., Goldblatt D., and Gennery A.R. 2013. Clinical manifestations of disease in X-linked carriers of chronic granulomatous disease. J. Clin. Immunol. 33: 1276–1284.
Cale C.M., Jones A.M., and Goldblatt D. 2000. Follow up of patients with chronic granulomatous disease diagnosed since 1990. Clin. Exp. Immunol. 120: 351–355.
Connelly J.A., Marsh R., Parikh S., and Talano J.A. 2018. Allogeneic hematopoietic cell transplantation for chronic granulomatous disease: Controversies and state of the art. J. Pediatric Infect. Dis. Soc. 7: S31–S39.
De Ravin S.S., Naumann N., Cowen E.W., Friend J., Hilligoss D., Marquesen M., Balow J.E., Barron K.S., Turner M.L., Gallin J.I., and Malech H.L. 2008. Chronic granulomatous disease as a risk factor for autoimmune disease. J. Allergy Clin. Immunol. 122: 1097–1103.
Guide S.V., Stock F., Gill V.J., Anderson V.L., Malech H.L., Gallin J.I., and Holland S.M. 2003. Reinfection, rather than persistent infection, in patients with chronic granulomatous disease. J. Infect. Dis. 187: 845–853.
Ho C.M., Vowels M.R., Lockwood L., and Ziegler J.B. 1996. Successful bone marrow transplantation in a child with X-linked chronic granulomatous disease. Bone Marrow Transplant. 18: 213–215.
Kaplan E.L., Laxdal T., and Quie P.G. 1968. Studies of polymorphonuclear leukocytes from patients with chronic granulomatous disease of childhood: Bactericidal capacity for streptococci. Pediatrics, 41: 591–599.
Kaufmann K.B., Chiriaco M., Siler U., Finocchi A., Reichenbach J., Stein S., and Grez M. 2014. Gene therapy for chronic granulomatous disease: Current status and future perspectives. Curr. Gene Ther. 14: 447–460.
Kohn D.B. 2010. Update on gene therapy for immunodeficiencies. Clin. Immunol. 135: 247–254.
Köker M.Y., Camcıoğlu Y., van Leeuwen K., Kılıç S.Ş., Barlan I., Yılmaz M., Metin A., de Boer M., Avcılar H., Patıroğlu T., Yıldıran A., Yeğin O., Tezcan I., Sanal Ö., and Roos D. 2013. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J. Allergy Clin. Immunol. 132: 1156–1163.e5.
Kuhns D.B., Alvord W.G., Heller T., Feld J.J., Pike K.M., Marciano B.E., Uzel G., DeRavin S.S., Priel D.A., Soule B.P., Zarember K.A., Malech H.L., Holland S.M., and Gallin J.I. 2010. Residual NADPH oxidase and survival in chronic granulomatous disease. N. Engl. J. Med. 363: 2600–2610.
Lazarus G.M. and Neu H.C. 1975. Agents responsible for infection in chronic granulomatous disease of childhood. J. Pediatr. 86: 415–417.
Mahdaviani S.A., Mohajerani S.A., Rezaei N., Casanova J.L., Mansouri S.D., and Velayati A.A. 2013. Pulmonary manifestations of chronic granulomatous disease. Expert Rev. Clin. Immunol. 9: 153–160.
Marciano B.E., Rosenzweig S.D., Kleiner D.E., Anderson V.L., Darnell D.N., Anaya-O’Brien S., Hilligoss D.M., Malech H.L., Gallin J.I., and Holland S.M. 2004. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics, 114: 462–468.
Marciano B.E., Zerbe C.S., Falcone E.L., Ding L., DeRavin S.S., Daub J., Kreuzburg S., Yockey L., Hunsberger S., Foruraghi L., Barnhart L.A., Matharu K., Anderson V., Darnell D.N., Frein C., Fink D.L., Lau K.P., Long Priel D.A., Gallin J.I., Malech H.L., Uzel G., Freeman A.F., Kuhns D.B., Rosenzweig S.D., and Holland S.M. 2018. X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J. Allergy Clin. Immunol. 141: 365–371.
Matute J.D., Arias A.A., Wright N.A., Wrobel I., Waterhouse C.C., Li X.J., Marchal C.C., Stull N.D., Lewis D.B., Steele M., Kellner J.D., Yu W., Meroueh S.O., Nauseef W.M., and Dinauer M.C. 2009. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood, 114: 3309–3315.
Murguia-Favela L. and Manson D. 2014. Imaging in patients with chronic granulomatous disease. LymphoSign J. 1: 105–120.
Roos D. and de Boer M. 2014. Molecular diagnosis of chronic granulomatous disease. Clin. Exp. Immunol. 175: 139–149.
Schappi M.G., Jaquet V., Belli D.C., and Krause K.H. 2008. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin. Immunopathol. 30: 255–271.
Seger R.A., Gungor T., Belohradsky B.H., Blanche S., Bordigoni P., Di Bartolomeo P., Flood T., Landais P., Müller S., Ozsahin H., Passwell J.H., Porta F., Slavin S., Wulffraat N., Zintl F., Nagler A., Cant A., and Fischer A. 2002. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: A survey of the European experience, 1985–2000. Blood, 100: 4344–4350.
Thomsen I.P., Smith M.A., Holland S.M., and Creech C.B. 2016. A comprehensive approach to the management of children and adults with chronic granulomatous disease. J. Allergy Clin. Immunol. Pract. 4: 1082–1088.
Towbin A.J. and Chaves I. 2010. Chronic granulomatous disease. Pediatr. Radiol. 40: 657–668; quiz 792–793.
Winkelstein J.A., Marino M.C., Johnston R.B. Jr., Boyle J., Curnutte J., Gallin J.I., Malech H.L., Holland S.M., Ochs H., Quie P., Buckley R.H., Foster C.B., Chanock S.J., and Dickler H. 2000. Chronic granulomatous disease: Report on a national registry of 368 patients. Medicine, 79: 155–169.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 7 • Number 2 • June 2020
Pages: 66 - 80
History
Received: 10 January 2020
Accepted: 24 February 2020
Accepted manuscript online: 19 April 2020
Copyright
© 2020.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
PariaKashani, LaraFarras Roca, and DavidManson. 2020. Radiographic features of patients with chronic granulomatous disease. LymphoSign Journal.
7(2): 66-80. https://doi.org/10.14785/lymphosign-2020-0002
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
