A case of common variable immune deficiency with lung disease–not just bronchiectasis
Abstract
Introduction: Common Variable Immune Deficiency (CVID) is the most prevalent form of severe antibody deficiency in children and adults. Most patients suffer recurrent, mainly sinopulmonary, infections. Despite adequate IVIG replacement therapy, chronic lung disease continues to be a main cause of morbidity and mortality. The term granulomatous-lymphocytic interstitial lung disease (GLILD) is frequently used to describe interstitial lung disease associated with immune dysregulation in primary antibody deficiency, such as CVID.
Aim: To describe the case of a 10-year-old male with CVID who developed GLILD and his response to treatment with Rituximab.
Discussion: Our patient is a young male with CVID and no genetic diagnosis, whose lung functions and general condition continued to deteriorate despite adequate intravenous immunoglobulin replacement therapy and mycophenolate mofetil treatment. After the diagnosis of GLILD, we initiated treatment with a 4-dose weekly course of Rituximab with prompt resolution of his interstitial disease. Although GLILD is a well described condition that accompanies CVID as a manifestation of immune dysregulation, it is still under recognized, especially in the pediatric population. Among experts, there is little uniformity when it comes to diagnostic and treatment approaches. Recent studies showed improved outcomes when using combination therapy with Rituximab, such as in our patient.
Statement of Novelty: We shed light on GLILD, an important condition that accompanies CVID, and demonstrate an excellent response to the steroid sparing agent Rituximab. This is a crucial aspect when considering therapeutic choices for the pediatric population.
Introduction
Common Variable Immune Deficiency (CVID) is the most prevalent form of severe antibody deficiency in children and adults. It is not a single disease but rather a collection of hypogammaglobinemia syndromes resulting from many genetic defects. Despite great advances in genetic sequencing and diagnosis in the last decades, in most cases, the cause is still unknown. CVID is defined by the following laboratory criteria: markedly reduced serum concentrations of immunoglobulin (Ig) G, in combination with low levels of IgA and/or immunoglobulin IgM, poor or absent response to immunizations, and an absence of any other defined immunodeficiency state. When these laboratory findings are accompanied by a relevant clinical phenotype, the diagnosis of CVID is confirmed.
Clinical phenotypes of CVID vary. Most patients have recurrent infections, mainly sinopulmonary infections. Other clinical features include lymphoproliferation, autoimmune cytopenia, and granulomatous disease (Bousfiha et al. 2020). The mainstay of treatment consists of antibiotic prophylaxis and IVIG replacement therapy (IGRT). In selected cases HSCT is considered. Despite adequate IGRT, chronic lung disease continues to cause morbidity and mortality in CVID patients.
Respiratory infections can cause structural lung damage that may promote chronic pulmonary disease. Delay in diagnosis is associated with fixed pulmonary obstruction, chronic atelectasis, pulmonary fibrosis, and bronchiectasis. Chronic lung disease may also develop because of immune dysregulation independent of infection or deficiencies in host defense that are not alleviated by IVIG or antibiotic prophylaxis (Maglione 2020). Interstitial lung disease develops in 10-20% of patients with CVID. The term granulomatous-lymphocytic interstitial lung disease (GLILD) is frequently used to describe the interstitial lung disease associated with primary antibody deficiency (Maglione 2020).
We hereby present a patient whose case highlights the importance of recognizing this condition and offer appropriate treatment.
Case Report
A 10-year-old male patient was referred to our immunology clinic in 2018. His prior medical history included atopic dermatitis, recurrent otitis media, and recurrent pneumonia. He presented with recurrent fever and a productive cough. He had no relevant family history and was of Russian decent. His physical exam revealed lymphadenopathy and splenomegaly. Auscultation of the lungs revealed distinct rales.
His laboratory workup showed leukopenia, lymphopenia, and microcytic normochromic anemia. Shortly after initial presentation he also developed thrombocytopenia. His direct and indirect Coombs test was positive. He underwent a basic evaluation for ALPS (autoimmune lymphoproliferative syndrome) which demonstrated a slightly elevated population of DNT cells of 2.7% in addition to elevation of B12, IL-10, SOL–IL2, and FAS-L.
When examining his immunoglobulin levels, we could see that he had a profound antibody deficiency. His IgG levels were 185 (mg/dL), IGA–<6, IgM–21.1, IgE <4.54. He also had a poor immunological response to the vaccinations he received — tetanus, diphtheria, strep. Pneumonia (Table 1). His lymphocyte subpopulations phenotype was normal.
Table 1:

Imaging included a chest X-ray which was prominent for interstitial infiltrates, left lower lobe infiltrate and hilar enlargement. CT scan demonstrated the typical radiological pattern of bronchiectasis in addition to bilateral enlarged hilar lymph nodes, enlarged mediastinal & axillar lymph nodes and a subcarinal mass in the posterior mediastinum, enveloping the main bronchi. There was splenomegaly (16 cm) on abdominal ultrasound. PET CT localized a “hot” axillar gland. A lymph node biopsy ruled out malignancy and demonstrated follicular hyperplasia. Spirometry showed a restrictive pattern with no reversibility, and normal lung volume. Diffusing capacity for carbon monoxide (DLCO) was normal. Bronchoscopy was consistent with chronic airway inflammation. A working diagnosis of CVID was established and he was started on IVIG replacement therapy. He also received azithromycin prophylaxis. Whole exome sequencing did not reveal a recognized genetic disorder.
A year after first presenting to our clinic he developed worsening pancytopenia. Due to his worsening condition, we added mycophenolate mofetil (MMF) to his treatment regimen. He remained cytopenic but with stable counts and his lymphadenopathy lessened.
Unfortunately, due to poor compliance the family stopped treatment with MMF. He presented in 2020 with weight loss and worsening respiratory symptoms. CT demonstrated worsening of all previous findings, further enlargement of the spleen, and diffuse lymphadenopathy (Figure 1). There was a decline in total lung capacity on spirometry and also his DLCO was diminished. His immunoglobulin levels were stable. He had pancytopenia (leukopenia 1000–2000, lymphopenia 500–900, neutropenia 400–600, Hb 10.5–12, PLT 75K). Flow cytometry showed low B cells (CD19–2.5%, CD20–2.5%), and absence of memory B cells (CD27+CD19−0%) (Table 1). Due to his dramatic clinical decline, we proceeded with a lung biopsy. Pathology showed diffuse lymphoid hyperplasia (Figure 2) with positive immunostains for CD3 and CD20. This is consistent with a diagnosis of granulomatous lymphocytic interstitial lung disease (GLILD).
Figure 1:
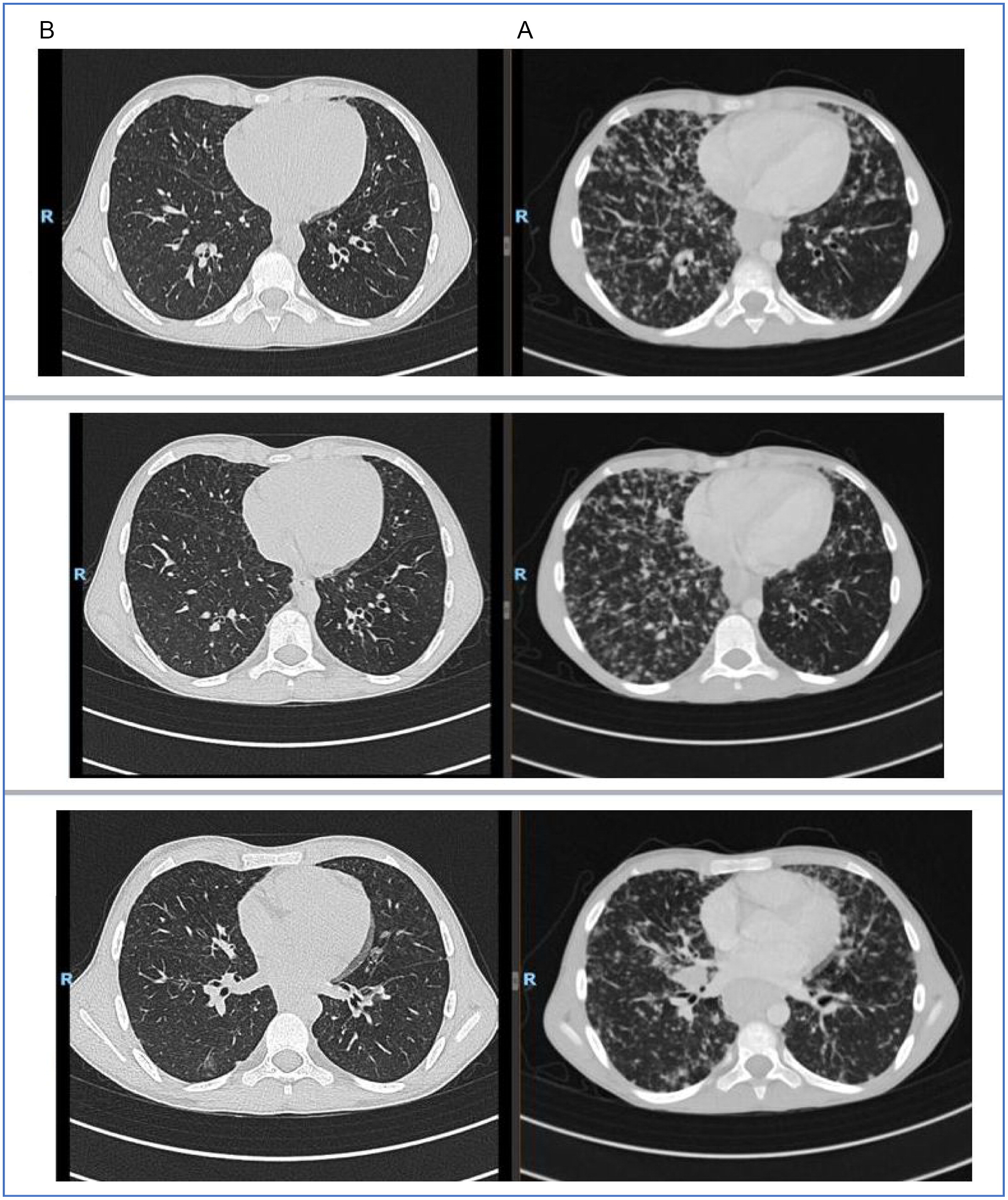
Figure 2:

Due to the presence of both B and T cells in the lung infiltrate of GLILD patients, we added a 4-dose course of Rituximab (dose 1 week apart) to his treatment and also optimized his IGRT to achieve trough levels of at least a 1000 mg/dL. CT scan 2 months after completion of Rituximab therapy showed nearly complete remission of lung disease with residual findings of his bronchiectatic lesions (Figure 1). His lung functions significantly improved.
Discussion
Although GLILD, a chronic lung disease which is a manifestation of the immune dysregulation that accompanies CVID is well described, it is still under recognized especially in the pediatric population.
Alterations in T cell function, generalized immune dysregulation, greater infection susceptibility, and/or the presence of pathogenic B cells may be fundamental to the development of interstitial lung disease in primary antibody deficiency. The presenting symptoms and physical examination are non-specific and high-resolution computed tomography is vital for the evaluation. A lung biopsy confirms the diagnosis while ruling out malignancy. Pathology is typically consistent with one or more forms of benign pulmonary lymphoproliferation. Granulomatous inflammation and organizing pneumonia may also be found in lungs of patients with primary antibody deficiency together with one of the lymphoproliferative pathologies (Maglione 2020).
Predominant cells in the infiltrate are CD4+ T cells, but nodules of CD20+ B cells surrounded by CD4+ T cells are also found, mainly localized to the interstitium. Regulatory T cells are absent in the lungs in GLILD (Baumann et al. 2018).
In patients with CVID, interstitial lung disease frequently occurs in conjunction with lymphoid hyperplasia in other tissues, such as in our patient. The pulmonary lymphoid hyperplasia that characterizes the interstitial lung disease seen in primary antibody deficiency may reflect systemic immune dysregulation inherent to the patient (Maglione 2020).
The European GLILD network (e-GLILDnet) aims to describe how GLILD is currently managed in clinical practice. They developed and conducted an online survey facilitated by the European Society for Immunodeficiencies (ESID) and the European Respiratory Society (ERS) between February and April 2020. There was little uniformity in diagnostic or therapeutic interventions.
Sixty-six percent used steroids for remission-induction and 47% for maintenance therapy. Azathioprine, Rituximab, and MMF were the most frequently prescribed steroid-sparing agents. Pulmonary function tests were the preferred modality for monitoring patients during follow-up (van de Ven et al. 2020). It should be noted that in a consensus statement from the British Lung Foundation, it is stated that expectant management is a viable option, and not to treat a patient who is asymptomatic with normal and stable lung function (Hurst et al. 2017).
In our case, the patient was symptomatic and had a decline in lung function as his status deteriorated. As such, we decided to add another immunosuppressive agent and he received a course of Rituximab. He continued his other treatments as well. Verbsky et al. (2021) demonstrated in a group of 39 patients who had CVID and GLILD that combination therapy with Rituximab improved lung function and CT scores. Especially in young children, it is important to have a reliable steroid sparing agent and this protocol shows very promising results.
Our patient showed prompt resolution of pulmonary findings after Rituximab therapy and we will continue to monitor long term treatment effects.
REFERENCES
Baumann U., Routes J.M., Soler-Palacín P., and Jolles S. 2018. The lung in primary immunodeficiencies: New concepts in infection and inflammation. Front. Immunol. 9: 1837.
Bousfiha A., Jeddane L., Picard C., Al-Herz W., Ailal F., Chatila T., Cunningham-Rundles C., Etzioni A., Franco J.L., Holland S.M., Klein C., Morio T., Ochs H.D., Oksenhendler E., Puck J., Torgerson T.R., Casanova J.L., Sullivan K.E., and Tangye S.G. 2020. Human inborn errors of immunity: 2019 Update of the IUIS phenotypical classification. J. Clin. Immunol. 40(1): 66–81.
Hurst J.R., Verma N., Lowe D., Baxendale H.E., Jolles S., Kelleher P., Longhurst H.J., Patel S.Y., Renzoni E.A., Sander C.R., Avery G.R., Babar J.L., Buckland M.S., Burns S., Egner W., Gompels M.M., Gordins P., Haddock J.A., Hart S.P., Hayman G.R., Herriot R., Hoyles R.K., Huissoon A.P., Jacob J., Nicholson A.G., Rassl D.M., Sargur R.B., Savic S., Seneviratne S.L., Sheaff M., Vaitla V.P.M., Walters G.I., Whitehouse J.L., Wright P.A., and Condliffe A.M. 2017. British lung foundation/united kingdom primary immunodeficiency network consensus statement on the definition, diagnosis, and management of GLILD in CVID. J. Allergy Clin. Immunol. Pract. 5(4): 938–945.
Maglione P.J. 2020. Chronic lung disease in primary antibody deficiency: Diagnosis and management. Immunol. Allergy Clin. North Am. 40(2): 437–459.
van de Ven A.A.J.M., Alfaro T.M., Robinson A., Baumann U., Bergeron A., Burns S.O., Condliffe A.M., Fevang B., Gennery A.R., Haerynck F., Jacob J., Jolles S., Malphettes M., Meignin V., Milota T., van Montfrans J., Prasse A., Quinti I., Renzoni E., Stolz D., Warnatz K., and Hurst J.R. 2020. Managing granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency disorders: E-glildnet international clinicians survey. Front Immunol. 11: 606333.
Verbsky J.W., Hintermeyer M.K., Simpson P.M., Feng M., Barbeau J., Rao N., Cool C.D., Sosa-Lozano L.A., Baruah D., Hammelev E., Busalacchi A., Rymaszewski A., Woodliff J., Chen S., Bausch-Jurken M., and Routes J.M. 2021. Rituximab and antimetabolite treatment of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency. J. Allergy Clin. Immunol. 147(2): 704–712.e17.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 8 • Number 3 • September 2021
Pages: 81 - 85
History
Received: 22 August 2021
Accepted: 30 August 2021
Accepted manuscript online: 30 August 2021
Copyright
© 2021.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
SharonPolakow Farkash, ShaiEhrlich, GuySteuer, JoanneYacobovich, NirDiamant, and NufarMarcus. 2021. A case of common variable immune deficiency with lung disease–not just bronchiectasis. LymphoSign Journal.
8(3): 81-85. https://doi.org/10.14785/lymphosign-2021-0026
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
