Leukocyte adhesion deficiency type II: A rare case of primary immune deficiency with unique finding of para-Bombay phenotype
Joshua Hochmana, Akash Guptab, Michelle Zellerc, Rae Bragera
aDivision of Clinical Immunology and Allergy, McMaster University, Hamilton, ON, Canada
bDepartment of Pathology and Molecular Medicine, McMaster University, Hamilton, ON, Canada
cDivision of Hematology and Thromboembolism, Department of Medicine, McMaster University, Hamilton, ON, Canada
Introduction: Leukocyte adhesion deficiency type II (LAD-II) is a rare autosomal recessive primary immunodeficiency, caused by absence in the Golgi GDP-fucose transport protein (GFTP). This protein is encoded by
SLC35C1 (solute carrier family 35 member C1) or
FUCT1 (GDP-fucose transporter 1) at 11p11.2 (OMIM *605881), in which 7 different mutations have been described thus far (
Van de Vijver et al. 2013). LAD-II was first reported in 1992, and since then less than 20 cases have been described worldwide. The initial cases were all clustered within a small geographical region in the Middle East, and pedigrees revealed an autosomal recessive inheritance (
Hanna and Etzioni 2012).
LAD-II has been categorized as one of the congenital disorders of glycosylation, and is also known as CDG-IIc. Patients with LAD-II have a defect in fucosylation of various cell-surface glycoproteins, such as sialyl Lewis X carbohydrate groups (sLeX, CD15a) on leukocyte proteins, alpha1-6-core fucosylated N-glycans on fibroblast proteins, and blood group antigen H on erythrocytes, known as the Bombay blood group phenotype (
Van de Vijver et al. 2013). The primary immune disturbance is a defect in the initial rolling of leukocytes over the endothelial vessel wall in areas of inflammation, usually mediated by contact between L-selectins on leukocytes and E- or P-selectins on the endothelium, with their respective sialated fucosyl ligands on opposite cells (
Van de Vijver et al. 2013). As a result, there is decreased leukocyte adhesion and recruitment to areas of infection, resulting in recurrent infections such as cold abscesses. Infectious complications in LAD-II are mild compared to LAD-I and LAD-III, as adhesion and transmigration via Beta-2 integrin is intact, which allows some level of neutrophil mobilization (
Etzioni and Tonetti 2000). There is an associated finding of neutrophilia, secondary to impaired cell trafficking, which tends to remain elevated during childhood and declines to the normal range at adolescence. Fucosylation is a key physiologic process for many other biological systems, and therefore LAD-II patients have been reported as having clinical findings including developmental and growth delay, and coarse facial features. As well, one half of patients have developed convulsions, cerebral atrophy, and features of Autism (
Hanna and Etzioni 2012).
Case Report: A 10 year old female was referred to our outpatient immunology clinic, after confirming a genetic diagnosis of LAD-II. According to the medical history, the patient was born at 41 weeks via spontaneous vaginal delivery at full term, with a birth weight of 3.771 kg. Remainder of growth parameters at birth were within normal limits, and there were no documented dysmorphisms at birth. There was no consanguinity. The family is Caucasian, of Dutch descent. There was no delayed separation of the umbilical cord. Family history was non-contributory and the patient has three siblings who are clinically unaffected. The history of infections included 2 episodes of uncomplicated acute otitis media during her first few years of life. The patient also had history of three episodes of recurrent periodontitis between the ages of 6 and 9. All of these were treated with oral antibiotics, and also required dental extraction. She received regular dental cleanings in the interim. There was no other history of recurrent infections. All childhood vaccinations including live vaccinations were given, with no adverse events noted.
Genetic investigations were initially ordered for symptom constellation including short stature, facial dysmorphisms, and developmental delay. Short stature was not noted until age 3; growth parameters at diagnosis included: height 121.5 cm (<1st centile), weight 26 kg (7th centile), and head circumference 52.5 cm (50–98 centile). At 8 years old her bone age was 5.5 years. On physical exam, the patient had hypoplasia of the supraorbital ridges, broad nasal bridge and a normal philtrum. She had deep set eyes, wide central incisors, and normal palate. She had an ear pit behind the left ear. She had widely spaced nipples. There was no digital webbing. Respiratory, cardiovascular and abdominal exam were within normal limits, with no organomegaly. Upper limbs showed full range of motion and bulking, and tapering fingers with increased extension angle. Lower limbs had normal range of motion, with deep set toe nails. Neurological and dermatological examinations were unremarkable.
The patient was developmental delayed in the domains of gross motor, fine motor, and speech during her first year of life, and was later diagnosed with Autism Spectrum Disorder with associated learning disability. Initial investigations performed included karyotype which was normal female (46, XX), as well as chromosomal microarray which was normal and female. There were no changes in the SHOX gene and Fragile X testing was negative. Whole exome sequencing was then performed which demonstrated two pathologic variants, consistent with a diagnosis of LAD-II or CDG-IIc (SLC35C1 c.503_505delTCT; p.F168del, heterozygous, paternally inherited SLC35C1 c.439C>T; p.R147C, heterozygous, maternally inherited).
Further immunological bloodwork was then sent which showed hemoglobin 143 g/L, leukocyte count 6.0 × 109/L, platelets 287 × 109/L, neutrophils 2.5 × 109/L, lymphocytes 3.0 × 109/L, eosinophils 0.1 × 109/L. Total IgG 6.08 g/L, IgA 1.56 g/L, IgM 1.21 g/L. Flow cytometry: absolute CD3 count 1.95 × 109/L (65%), CD3CD4 absolute 1.14 × 109/L (38%), CD3CD8 absolute count 0.75 × 109/L (25%), CD4/CD8 ratio 1.5.
Immunohematologic workup identified that the patient’s blood group was O positive. Red blood cell phenotyping for the H-antigen was negative, with no detectable anti-H antibody present in her plasma. The patient was also Le(a-b+). Her direct antiglobulin test (DAT) was negative, and there were no clinically significant antibodies detected in her plasma.
Discussion: We report a case of LAD-II, a rare primary immunodeficiency, in an 8 year old in Canada. Our patient presented with many of the clinical features previously reported in LAD-II. However, our patient did not have intrauterine growth restriction, or dysmorphic features at birth, and only later developed short stature as well as other physical findings in keeping with diagnosis of LAD-II as a school-aged child (
Hanna and Etzioni 2012). In terms of clinical findings of immunodeficiency, our patient had recurrent periodontitis, requiring tooth extractions. There was no other history of recurrent infection. As well, she did not display expected hematologic findings such as neutrophilia. This contributed to delay in initial investigations, and biochemical and genetic investigations did not confirm a diagnosis until age 10.
Recurrent periodontitis in LAD has been well established, with findings including pathological bone resorption and premature loss of primary and permanent teeth (
Hajishengallis and Moutsopoulos 2014). This process was initially thought to be due to lack of neutrophil surveillance and transmigration to the peripheral oral tissues and is seen in all types of LAD. In LAD-II, an otherwise phenotypically mild immune deficiency, recurrent periodontitis still occurs, without other recurrent infections due to impaired neutrophil chemotaxis, such as cold abscesses. Recent studies have proposed this periodontitis may represent dysregulated over-expression of bone-resorptive cytokine IL-17 (
Hajishengallis and Moutsopoulos 2016). The proposed homeostatic mechanism “neutrostat,” is activated when neutrophils cannot transmigrate to peripheral tissues, leading to unrestrained expression of IL-23, IL-17 and G-CSF. In patients with LAD-I, abundant overexpression of T cell drives IL-17 mRNA and protein expression in periodontal tissue. In a mouse model, antibody mediated antagonism was shown to prevent the periodontitis, implicating a biological role (
Hajishengallis and Moutsopoulos 2014). Overproduction of IL-17 in the peripheral tissues may also lead to bacterial overgrowth in the oral cavity, further driving this inflammatory process (
Hajishengallis and Moutsopoulos 2016). The proposed mechanism of inflammatory periodontitis, in contrast to uncontrolled infection, is consistent with our patient’s inadequate response to antibiotics, and resolution requiring mechanical extractions and surgical manipulation. The role of prophylactic antibiotics remains unclear in this setting.
The defect in fucosylation seen in LAD-II has also been described to affect the production of the H antigen on red blood cells, leading to a Bombay phenotype. Testing of this patient’s red blood cells was negative for the H-antigen, however; there were no detectable anti-H antibodies in her plasma. The patient was, however, found to be Le(a+b-), which indicates that she is producing the secretor (Se) gene and is able to form the H-antigen via fucosylation in her plasma. This phenotypic presentation is more consistent with a Para-Bombay phenotype, which has not previously been reported in a LAD-II patient. Patients who are unable to produce the H-antigen will typically form an anti-H antibody via various environmental exposures. Anti-H antibodies will cause acute hemolysis, and these patients must be transfused red blood cells from donors with the Bombay phenotype to avoid this outcome. Our patient presents with a unique phenotype where there is no evidence of an anti-H antibody, despite the defect in fucosylation. The patient may, therefore, be eligible to receive RBC transfusions from a larger selection of compatible donors, should the need arise.
Conclusion: We present a case of a child with LAD-II who has a history of short stature, dysmorphic facies, periodontal infections, Autism, and developmental delay. However, this patient did not present with IUGR, had no neutrophilia, and facial dysmorphisms did not become apparent until later in childhood. Furthermore, our patient has a unique, not previously reported, para-Bombay phenotype, which differentiates this presentation from other cases of LAD-II in the literature. Future study to investigate the functional defect leading to this clinical presentation may be warranted. This diagnosis should be considered in other children with recurrent dental infections who do not have all of the previously-described features of LAD-II.
REFERENCES
Etzioni, A., and Tonetti, M. 2000. Leukocyte adhesion deficiency II – from A to almost Z. Immunol. Rev. 178(1):138–147. PMID: 1213799. doi: 10.1034/j.1600-065x.2000.17805.x.
Hajishengallis, G., and Moutsopoulos, N.M. 2014. Etiology of leukocyte adhesion deficiency-associated periodontitis revisited: Not a raging infection but a raging inflammatory response. Expert Rev. Clin. Immunol. 10(8):973–975. PMID: 24931458. doi: 10.1586/1744666x.2014.929944.
Hajishengallis, G., and Moutsopoulos, N.M. 2016. Role of bacteria in leukocyte adhesion deficiency-associated periodontitis. Microb. Pathog. 94:21–26. PMID: 26375893. doi: 10.1016/j.micpath.2015.09.003.
Hanna, S., and Etzioni, A. 2012. Leukocyte adhesion deficiencies. Ann. N. Y. Acad. Sci. 1250(1):50–55. doi: 10.1111/j.1749-6632.2011.06389.x.
Van de Vijver, E., van den Berg, T.K., and Kuijpers, T.W. 2013. Leukocyte adhesion deficiencies. Hematol. Oncol. Clin. North Am. 27(1): 101–116. PMID: 23351991. doi: 10.1016/j.hoc.2012.10.001.
Heterozygous variants in FOXN1 among patients with abnormal T-cell receptor excision circles and lymphopenia: a single-centre experience
Ori Scotta, Amarilla Mandolaa, Yehonatan Pasternaka, Yael Dinur-Schejterb, Brenda Reida, Vy Hong-Diep Kima, Chaim M. Roifmana,c
aDivision of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
bBone Marrow Transplantation Department, Hadassah-Hebrew University Medical Center, Jerusalem, Israel
cCanadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and Research Institute, Toronto, ON, Canada
Introduction: The Forkhead box (FOX) superfamily comprises of a wide array of transcription factors, with key roles in tissue development and homeostasis (
Lam et al. 2013). Within the FOX family, FOX protein N1 (FOXN1) is a key regulator of thymic epithelium and skin development. FOXN1 dictates gene expression involved in thymic epithelial cell (TEC) differentiation, lymphoid progenitor migration from the bone marrow, antigen processing and thymocyte selection (
Nowell et al. 2011;
Romano et al. 2013;
Žuklys et al. 2016). Complete FOXN1 deficiency was first identified in mice displaying the nude/severe combined immunodeficiency (SCID) phenotype, consisting of congenital alopecia totalis, nail dystrophy and thymic aplasia (
Flanagan 1966). An equivalent phenotype was later described in humans harbouring biallelic loss-of-function (LOF) mutations in the
FOXN1 gene (
Pignata et al. 1996;
Frank et al. 1999). In contrast to the well-established phenotype seen with complete
FOXN1 LOF, the role of heterozygous
FOXN1 mutations in human immunity has been slower to emerge. In a recent case series, Bosticardo et al. describe for the first time a cohort of children with heterozygous
FOXN1 mutations, who were found to have low T-cell receptor excision circles (TREC) levels and/or lymphopenia (
Bosticardo et al. 2019). To further expand the knowledge basis regarding this novel entity, we herein describe our experience with 4 children identified by newborn screen (NBS) with low TREC levels and T-lymphopenia, all of whom harbour monoallelic mutations in the
FOXN1 gene.
Case Series
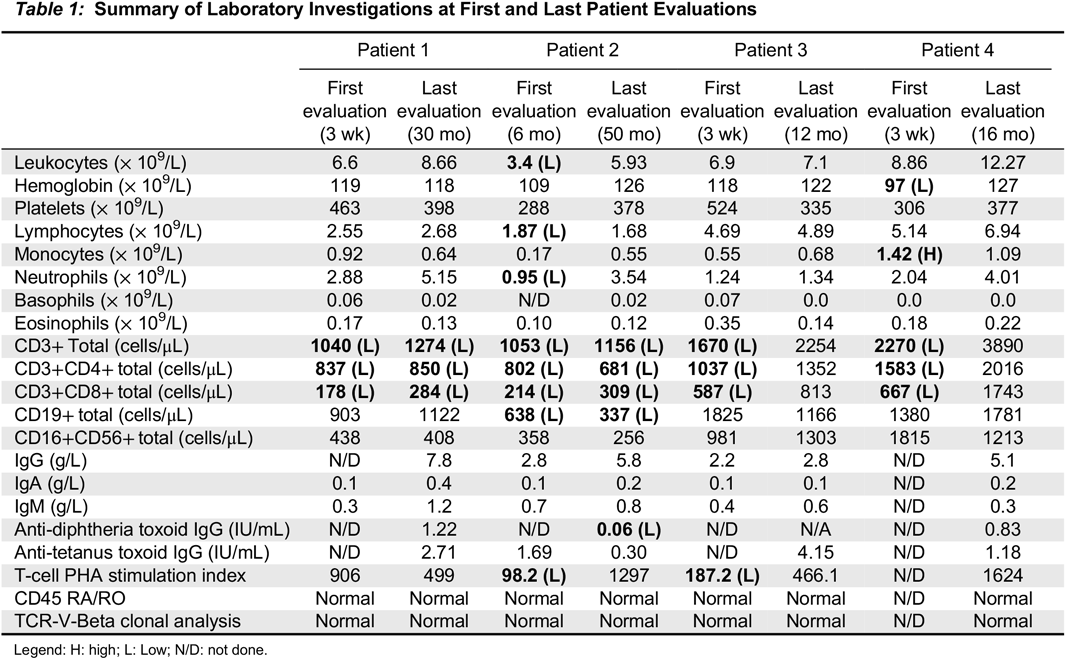
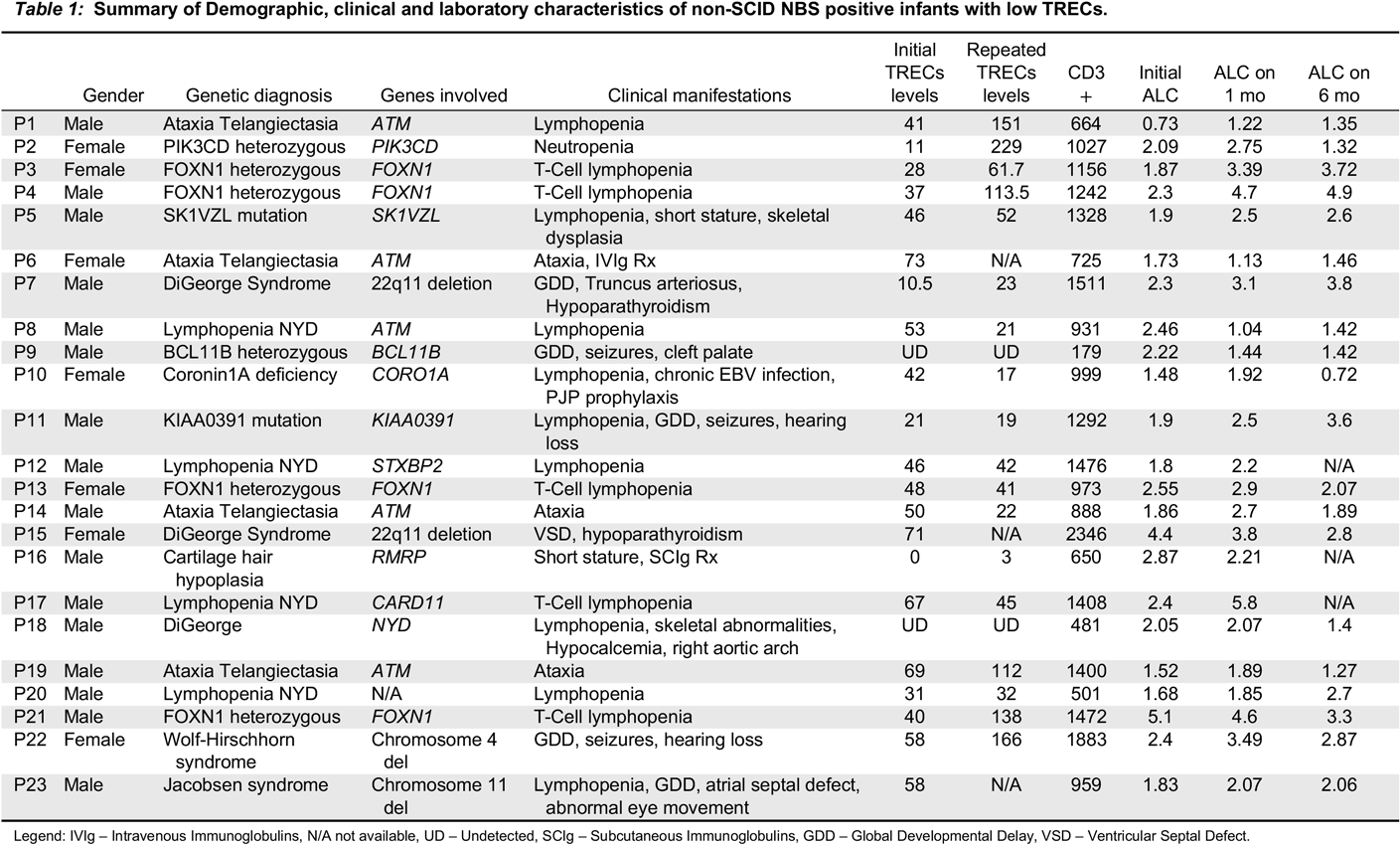
Case 1: An asymptomatic female newborn was referred for an abnormal TREC level identified on NBS testing from dried blood spot. She had been born at term (39 + 2 weeks) via uncomplicated spontaneous vaginal delivery following an unremarkable pregnancy. Family history identified non-consanguineous parents of mixed English, Dutch and African-American ancestry. Father had required tonsillectomy and adenoidectomy as a child for recurrent tonsillitis, and had a number of acute otitis media infections until the age of 4 years, for which he did not require any surgical intervention. Mother was healthy. Two maternal half siblings were healthy as well. There was no extended family history of immunodeficiency, autoimmunity, inflammation or early-onset malignancy. When seen at our clinic at the age of 3 weeks the girl had been growing and feeding well and had no notable concerns for infection or inflammation. Her physical exam was within normal limits. Laboratory investigations were notable for CD4 and CD8 lymphopenia, with normal B and NK cell counts; this persisted over the following 30 months. Functional assessment showed good humoral function noted by total immunoglobulins and killed-vaccine responses. Her T-cell mitogen proliferation index has always been normal. A summary of laboratory investigations on first and most recent assessment is available in
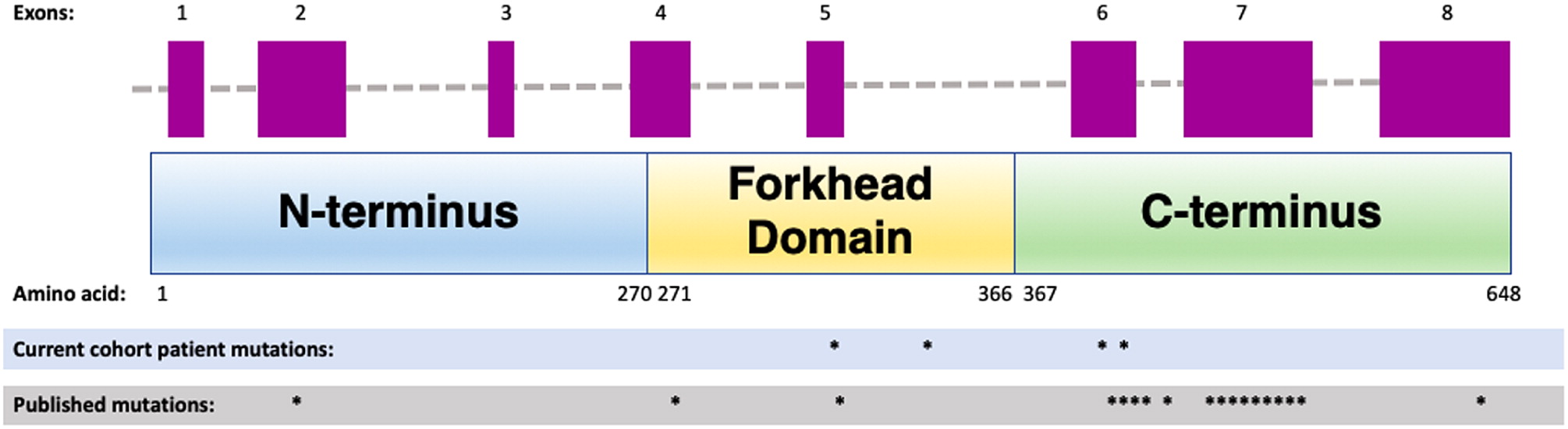
Table 1. Whole exome sequencing has revealed a novel heterozygous mutation in the forkhead domain of the
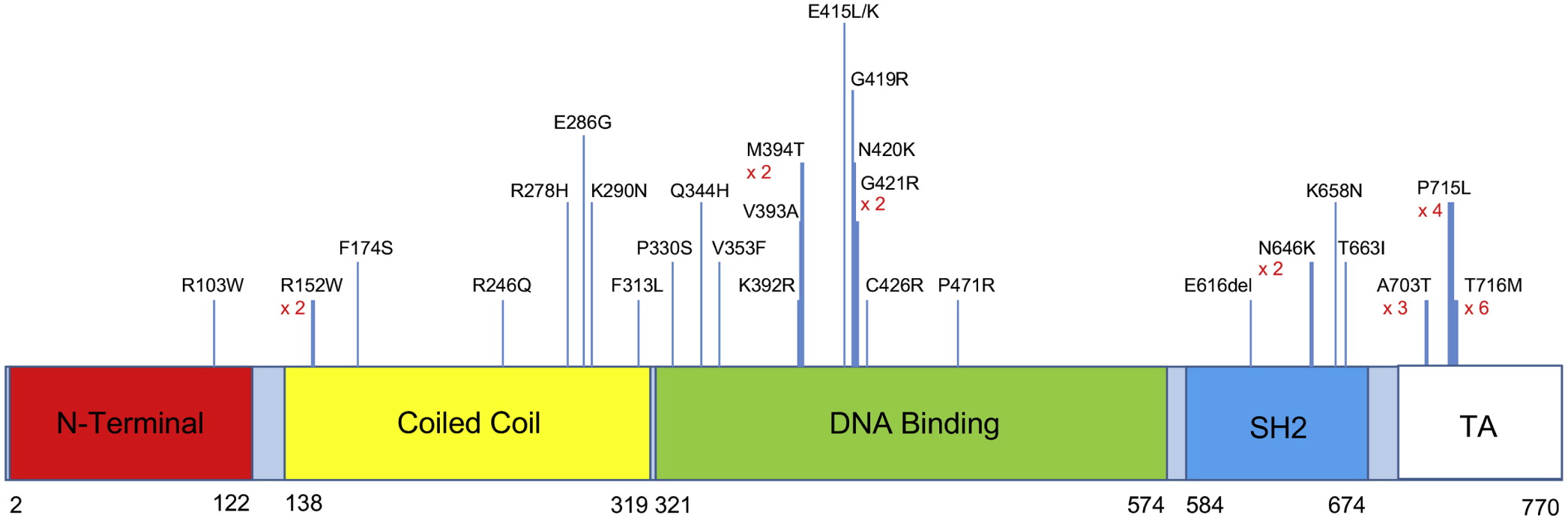
FOXN1 gene, disrupting an obligatory donor splice site. For a schematic graphic summary of patient mutations compared to previously published mutations in
FOXN1, please refer to
Figure 1.
Current clinical status: at present, the patient is 3 years old and remains clinically asymptomatic.
Case 2: An infant was referred to us at the age of 6 months for follow-up of abnormal TREC, lymphopenia, and recurrent respiratory infections. She was born at 38 weeks’ gestation following an unremarkable pregnancy and delivery, to non-consanguineous parents of Italian descent. Mother had a history of eczema and a thyroid goiter, with further maternal family members affected by Hashimoto’s thyroiditis, eczema, and celiac disease. The father was healthy. When seen for the first time, the patient had had two upper respiratory tract infections, and one pneumonia treated with oral antibiotics. Over the next 4 years she continued to have 3–4 upper respiratory tract infections per year as well as one Influenza infection requiring admission to hospital for Oseltamivir treatment. Concurrently, she was also diagnosed with asthma and eczema. She never had any deep-seated, fungal, viral, or opportunistic infections, and no further bacterial infections apart from the pneumonia at 5 months of age. Her physical examination when well never revealed any abnormalities except for eczema. Laboratory investigations (summarized in
Table 1) demonstrated persistent T-lymphopenia affecting both CD4 and CD8. She intermittently was found to have mild CD19 lymphopenia, although this sometimes coincided with recovery from viral respiratory infections. Her T-cell stimulation index to PHA was initially low, but has normalized over time. Her responses to killed vaccines were initially adequate, but have been declining over time. Whole genome sequencing was significant for a novel heterozygous missense variant in the forkhead domain of
FOXN1 (
Figure 1).
Current clinical status: Presently, at the age of 5 years the patient is well-grown and thriving, with occasional mild upper respiratory tract infections.
Case 3: This asymptomatic male infant was referred at the age of 3 weeks for an abnormal TREC level. He had been born at 40+5 weeks following an unremarkable pregnancy and spontaneous vaginal delivery. His parents were non-consanguineous of British/German/Russian/Romanian descent, and family history was significant for paternal grandmother with type 1 diabetes mellitus and hypothyroidism. Clinically, the boy has been well and has never developed any infectious, inflammatory or autoimmune manifestations. He has mild eczema on exam, for which he was treated with a moisturizer. His laboratory evaluation in early infancy was remarkable for CD4 and CD8 lymphopenia, as well as low T-cell proliferation response to PHA. Both T-cell numbers and function have normalized entirely by 1 year of life. He has never displayed any humoral abnormalities (
Table 1). SCID gene panel has demonstrated a heterozygous missense mutation in
FOXN1 affecting the forkhead domain, which has been previously described as pathogenic in the context of biallelic loss of function (
Figure 1).
Current clinical status: The patient is currently 4 years old, has received all age appropriate live-viral vaccines, and has had no further clinical concerns.
Case 4: A male infant was referred to us at 3 weeks of life given a finding of low TREC on NBS. He had been born at 37 weeks via spontaneous vaginal delivery. Pregnancy was the product of in-vitro fertilization, and was otherwise only remarkable for maternal inhaled corticosteroid use for asthma, with no oral corticosteroids required. Parents were of Ashkenazi Jewish descent and non-consanguineous, and family history was remarkable for asthma and eczema in both mother and older brother. Clinically, the boy has always been well with the exception of mild eczema. He has no infectious, inflammatory, or autoimmune manifestations. His laboratory investigations were initially remarkable for CD4 and CD8 lymphopenia in early infancy, which resolved over the next few months. His T-cell PHA stimulation index has been normal. His humoral assessment has always been within normal limits (
Table 1). Next generation sequencing in the form of a primary immunodeficiency gene panel showed a heterozygous, frameshift-inducing deletion in the forkhead domain of the
FOXN1 gene (
Figure 1).
Current clinical status: The patient is currently 2 years old, is well and thriving, and has begun receiving age appropriate live-viral vaccines. He has had no further clinical concerns.
Discussion:The implementation of NBS in Ontario, as well as in various provinces across Canada, has led to an increased detection rate of patients with non-SCID lymphopenia who display low TREC levels shortly after birth. In this regard, heterozygous FOXN1 mutations have been recently identified as a genetic etiology in a subset of children with abnormal NBS and/or lymphopenia. In our centre, 4 children affected by such mutations have demonstrated an overall reassuring clinical course, with mostly isolated T-cell lymphopenia which improves over time. Of note, the T-lymphopenia has never been in the severe range for any of our patients, and has never merited any intervention such as antimicrobial prophylaxis. Two of the children in our cohort showed evidence of abnormal T-cell function early on in infancy; this has normalized in both patients by the end of the first year of life, and was not accompanied by infections typical of SCID patients. One patient in our cohort, who presented with frequent respiratory infections, further displayed evidence of humoral abnormalities reflected in her fluctuating CD19 counts and vaccine responses. It is possible that this patient has an additional immune abnormality which has not yet been identified, beyond the FOXN1 mutation.
A recent case series has outlined a multi-centre experience caring for 25 children and 22 adults with heterozygous
FOXN1 mutations (
Bosticardo et al. 2019). Of the children, 21 were identified following an abnormal NBS result, while 4 others were investigated for persistent severe lymphopenia and/or recurrent infections. Most were clinically well except for viral respiratory infections, and were managed conservatively. Common non-infectious manifestations included eczema and nail dystrophy. Five patients displayed evidence of more profound lymphopenia and/or severe infections, and three received a hematopoietic stem cell transplant (HSCT) for a clinical diagnosis of SCID, before their
FOXN1 mutations had been identified. Unfortunately, transplant outcome has included persistent lymphopenia despite engraftment in two children, and mortality of infection in the third patient. With respect to adults included in the report, the majority did not report on any substantial recurrent infectious history; however, it is possible that individuals impacted by severe infections would not have survived into adulthood, thus
de-facto excluding persons with more profound disease manifestations from the adult cohort. In terms of laboratory evaluation, the most common and consistent manifestation in infants and children was T-lymphopenia impacting both CD4 and CD8 populations, with improvement in CD4 noted toward the second year of life. Among adult participants, CD8 lymphopenia was the only persistent disease abnormality.
The spectrum of clinical manifestations and severity hitherto reported among children with heterozygous FOXN1 mutations has been broad, with no clear genotype-phenotype correlation with respect to location and nature of the mutations. Indeed, mutations reported by Bosticardo et al. spanned all gene domains, and included substitutions, insertions and deletions, in both exonic and intronic regions. While 2 out of 3 children who underwent HSCT were affected by frameshift-inducing mutations, other children harbouring such mutations were reported to be asymptomatic. Of note, the authors did not determine whether gene variants resulted in an absent gene product or a dominant negative truncated protein, which could potentially account for the variable presentations.
Patients with abnormal TREC and lymphopenia, present the clinician with a number of challenges with respect to frequency and nature of follow-up, extent of investigations required, and clinical management. In the case of children with heterozygous FOXN1 mutations, our centre has practiced conservative, yet diligent follow-up of patients with abnormal lymphocyte counts and/or function. Care should be taken to rule out other possible underlying causes of lymphopenia such as perinatal stressors, fetal/maternal exposures, congenital infections, and other primary immunodeficiencies. In regards to live vaccines, an individually-tailored approach should be employed. Our experience suggests that in children with FOXN1 mutations and normal T-cell function, whose T-lymphopenia is mild and improving or fully resolved, live viral vaccinations may be administered safely. However, a case-by-case discussion should be held with families, identifying the potential risks and benefits of such an approach. For asymptomatic children whose laboratory values have normalized, annual follow-up is recommended to ensure no new manifestations have developed, and that immunological tests are stable. Close attention should also be given to dermatological manifestations which may develop over time should be. Finally, genetic testing and counselling may be considered for asymptomatic first-degree family members as well. We further encourage clinicians to report their findings, so we may globally expand our knowledge of this entity, providing better care and counselling to patients and families.
REFERENCES
Bosticardo, M., Yamazaki, Y., Cowan, J., Giardino, G., Corsino, C., Scalia, G., Prencipe, R., Ruffner, M., Hill, D.A., Sakovich, I., and Yemialyanava, I. 2019. Heterozygous FOXN1 variants cause low TRECs and severe T cell Lymphopenia, revealing a crucial role of FOXN1 in supporting early Thymopoiesis. Am. J. Hum. Genet. 105(3):549–561. PMID: 31447097. doi: 10.1016/j.ajhg.2019.07.014.
Flanagan, S.P. 1966. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet. Res. 8(3):295–309. PMID: 5980117. doi: 10.1017/S0016672300010168.
Frank, J., Pignata, C., Panteleyev, A.A., Prowse, D.M., Baden, H., Weiner, L., Gaetaniello, L., Ahmad, W., Pozzi, N., Cserhalmi-Friedman, P.B., and Aita, V.M. 1999. Exposing the human nude phenotype. Nature. 398(6727):473–474. PMID: 10206641. doi: 10.1038/18997.
Lam, E.W., Brosens, J.J., Gomes, A.R., and Koo, C.Y. 2013. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer. 13(7):482–495. PMID: 23792361. doi: 10.1038/nrc3539.
Nowell, C.S., Bredenkamp, N., Tetelin, S., Jin, X., Tischner, C., Vaidya, H., Sheridan, J.M., Stenhouse, F.H., Heussen, R., Smith, A.J., and Blackburn, C.C. 2011. Foxn1 regulates lineage progression in cortical and medullary thymic epithelial cells but is dispensable for medullary sublineage divergence. PLoS Genet. 7(11):e1002348. PMID: 22072979. doi: 10.1371/journal.pgen.1002348.
Pignata, C., Fiore, M., Guzzetta, V., Castaldo, A., Sebastio, G., Porta, F., and Guarino, A. 1996. Congenital alopecia and nail dystrophy associated with severe functional T‐cell immunodeficiency in two sibs. Am. J. Med. Genet. 65(2):167–170. PMID: 8911612. doi: 10.1002/(SICI)1096-8628(19961016)65:2<167::AID-AJMG17>3.0.CO;2-O.
Romano, R., Palamaro, L., Fusco, A., Giardino, G., Gallo, V., Del Vecchio, L., and Pignata, C. 2013. FOXN1: A master regulator gene of thymic epithelial development program. Front. Immunol. 4:187. PMID: 23874334. doi: 10.3389/fimmu.2013.00187.
Žuklys, S., Handel, A., Zhanybekova, S., Govani, F., Keller, M., Maio, S., Mayer, C.E., The, H.Y., Hafen, K., Gallone, G., and Barthlott, T. 2016. Foxn1 regulates key target genes essential for T cell development in postnatal thymic epithelial cells. Nat. Immunol. 17(10):1206–1215. PMID: 27548434. doi: 10.1038/ni.3537.
Prolidase deficiency: description of its presentation and detailed immunological assessment
Nora Alrumayyana, Sarah McAlpinea, Thomas Issekutza, Drew Slauenwhitea, Adam M. Huberb, Zaiping Liuc, Beata Derfalvia
aDivision of Immunology, Dalhousie University, IWK Health Centre, Halifax, NS, Canada
bDivision of Rheumatology, Department of Paediatrics, Dalhousie University, IWK Health Centre, Halifax, NS, Canada
cDivision of Clinical Biochemistry & Maritime Newborn Screening, Department of Pathology and Laboratory Medicine, Dalhousie University, IWK Health Centre, Halifax, NS, Canada
Background: Prolidase deficiency (PD) is an autosomal recessive inborn error of metabolism caused by mutations in the
PEPD gene encoding the enzyme prolidase D, leading to a defect in collagen synthesis (
Goodman et al. 1968;
Bousfiha et al. 2020). Although the International Union of Immunological Societies classifies PD under the immune dysregulation group, within the inborn errors of immunity, a detailed immunological assessment has never been reported (
Kelly et al. 2010;
Kurien et al. 2013). PD is also characterized as a “lupus-mimic” (
Shrinath et al. 1997;
Costa-Reis and Sullivan 2017), presenting predominantly with cutaneous features but also with systemic autoimmunity. Typical presentations include intractable skin ulceration, telangiectasia, recurrent infections, splenomegaly and dysmorphic features with intellectual disabilities. Thrombocytopenia, hypocomplementemia and hypergammaglobulinemia are frequent laboratory findings, as well as increased proline metabolites in serum and urine (
Freij and der Kaloustian 1986;
Dunn et al. 2011).
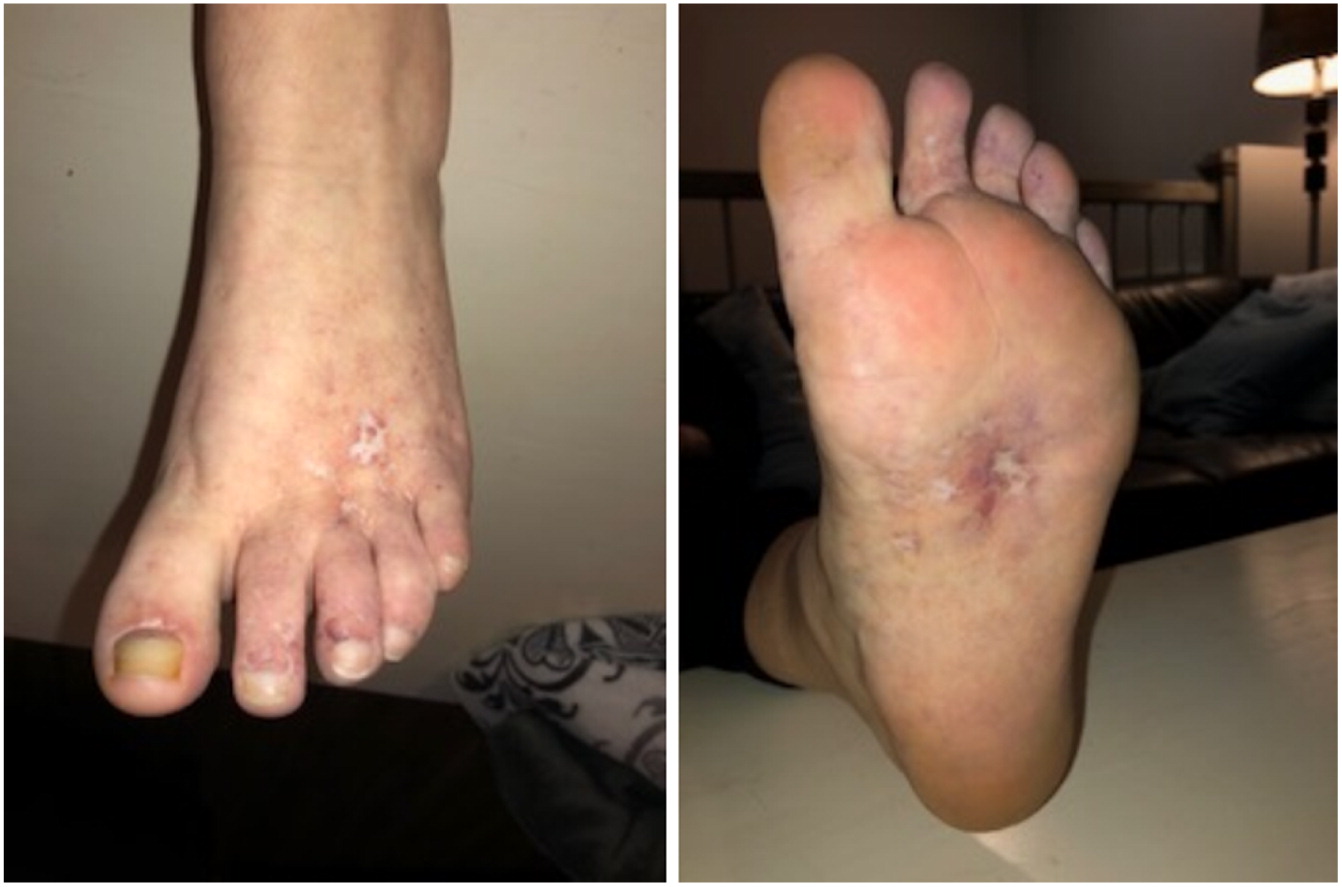
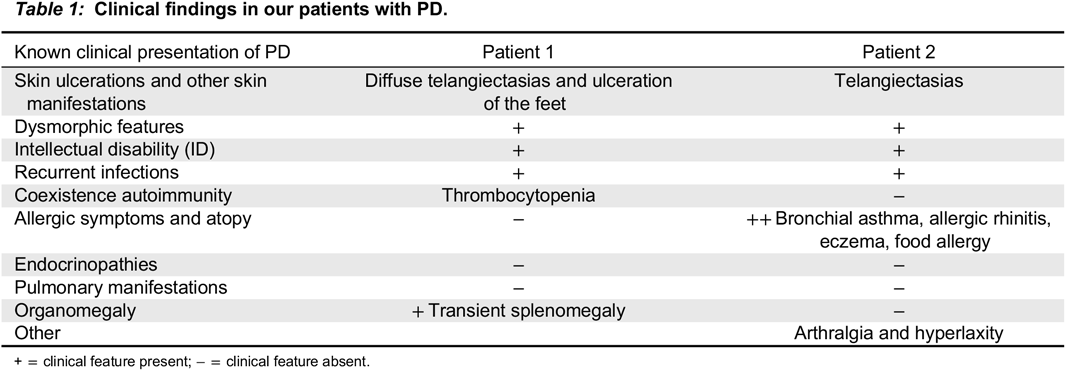
Case Description: We report identical twin sisters with PD and with variable severity of the phenotype. The index case, Patient 1, was a 15 year old female who had required a tonsillectomy, adenoidectomy, and bilateral myringotomy tubes for recurrent ear and throat infections in early childhood. This reduced the frequency of infections but did not completely eliminate them. At age 13 years, she presented with severely painful, chronic, refractory, oozing skin ulcerations on her lower extremities, which prevented ambulation so she needed to use a wheelchair for a year. On physical exam she had dysmorphic features including a low hairline, mild ptosis, hypertelorism, a depressed nasal root, a beak-like nose, and micrognathia. She had diffuse telangiectasias and ulcers on her feet (
Figure 1). A skin biopsy did not show a vasculitis, but instead a livedoid vasculopathy with perivascular lymphocytic infiltration. When she was 15 years of age, a Primary Immunodeficiency gene panel revealed two novel pathogenic compound heterozygous mutations (c.977G>A, p.(Trp326*) and c.550C>T; p.(Arg184*), both premature stop codons) in the
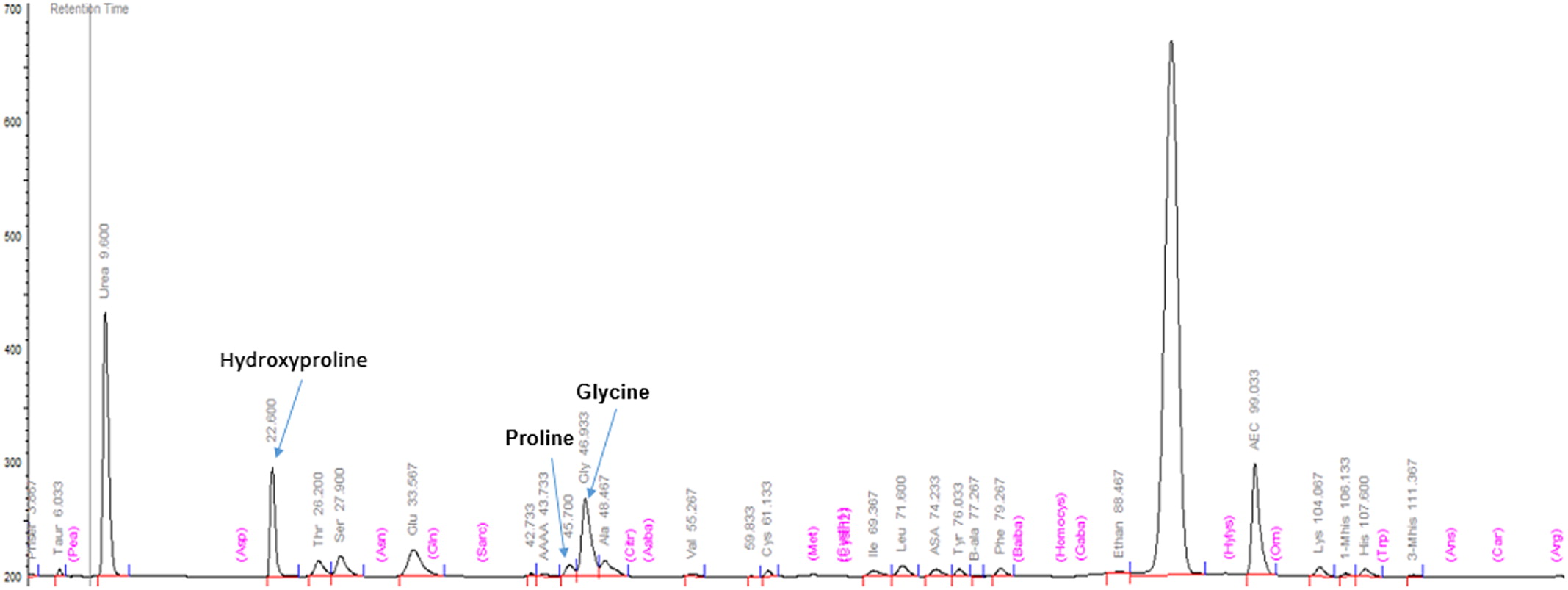
PEPD gene responsible for PD. The non-consanguineous parents are each carriers of one of the mutations, but are otherwise healthy. Massive imidodipeptiduria indicating increased proline metabolites also confirmed PD (
Figure 2).
Patient 1 initially seemed to respond to symptomatic therapies including high dose of intravenous immune globulin (IVIG), low molecular weight heparin, collagen synthesis cofactor vitamin C and a combination of topical tacrolimus (0.33%) and Proline-Glycine 5% ointment, allowing her to recover full physical activity. Unfortunately, this improvement was only temporary.
Patient 2 is the identical twin and had the same mutations. She also exhibited massive imidopeptiduria supporting the diagnosis of PD. She has the same typical facial features and similar telangiectasias to her sister, but with fewer respiratory tract infections and no skin ulceration. She has severe atopy. Clinical findings for both patients are summarized in
Table 1.
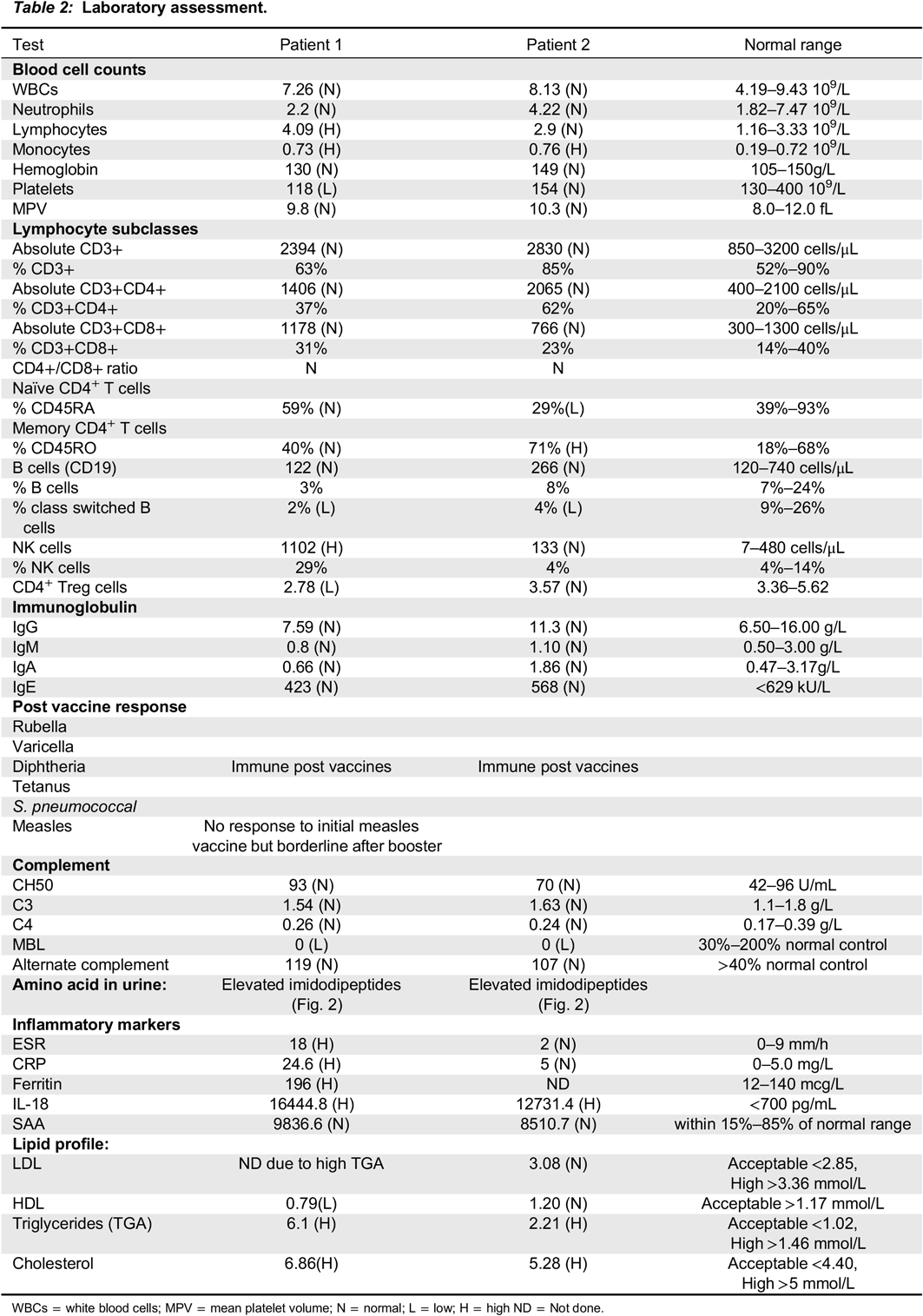
Methods: Informed consent was obtained. Detailed laboratory assessment included: measurement of immunoglobulin levels; antibody responses to various vaccines; classical, alternate and mannose binding lectin (MBL) complement activation with ELISA; and lymphocyte proliferation to mitogens and antigens (
Kurien et al. 2013) with H thymidine incorporation assay (
Table 2). Plasma inflammatory cytokine profile and serum amyloid A was measured using Multiplex Immunoassay. Natural killer (NK) cell cytotoxicity (degranulation and killing) and detailed immunophenotyping of T, B and NK lymphocyte subsets were obtained by flow cytometry. Urine amino acids were quantitated using a Biochrom amino acid analyzer.
Results: Immunoglobulin (IgG, IgG1-4, IgA, IgM, IgE) levels, antibody responses to vaccines and lymphocyte proliferation were normal. Both patients have MBL deficiency, which might contribute to the increased frequency of ear, nose, and throat infections. Autoantibody measurements including: ANA, ENA, Anti-TTG-IgA, Anti-cardiolipin, Anti-B2GP1, LA, ASMA, APCP, Anti-LKM, Anti-PR3, Anti-MPO, Anti-CCP and RF were all negative.
Lymphocyte subsets were normal, except both patients had high naïve B cells and low class switched memory B cells. Decreased Treg cell counts were found in Patient 1 with the more aggressive disease phenotype. Low NK cell killing was detected only in Patient 2. Blood inflammatory parameters (CRP, ESR) were elevated in Patient 2, who had more aggressive disease phenotype. SAA and several proinflammatory cytokines were normal in both patients, except plasma IL-18 level, which was extremely high (20-fold increase) compared to healthy controls. Serum lipid profile revealed high LDL and triglyceride in both patients, which has not been reported previously in PD (
Table 2).
Conclusion: We describe twin sisters with PD that is associated with abnormalities in immune function. Immunologic abnormalities may be a feature of PD, although this finding will require confirmation in additional patients. Our immunological assessment suggests that patients with PD may have a defect in T regulatory cells which seems to correlate to the severity of PD. Class switched memory B cells are reduced in number but the explanation for this is not clear. The extremely high IL-18 plasma levels suggest that PD might have an autoinflammatory component resulting from inflammasome activation. Our proposed new therapeutic regimen, which included immunomodulatory dose IVIG, microcirculation improving low molecular weight heparin and combination of the collagen synthesis cofactor vitamin C and topical proline, appeared to be resolve the skin ulceration and a significant improvement in quality of life.
REFERENCES
Bousfiha, A., Jeddane, L., Picard, C., Al-Herz, W., Ailal, F., Chatila, T., Cunningham-Rundles, C., Etzioni, A., Etzioni, J.L., Holland, S.M., Klein, C., Morio, T., Ochs, H.D., Oksenhendler, E., Puck, J., Torgerson, T.R., Casanova, J.L., Sullivan, K.E., and Tangye, S.G. 2020. Human inborn errors of immunity: 2019 Update of the IUIS Phenotypical classification. J. Clin. Immunol. 40(1):66–81. PMID: 32048120. doi: 10.1007/s10875-020-00758-x.
Costa-Reis, P., and Sullivan, K.E. 2017. Monogenic lupus: It’s all new! Curr. Opin. Immunol. 49:87–95. PMID: 29100097. doi: 10.1016/j.coi.2017.10.008.
Dunn, R., Varigos, G., and Winship, I. 2011. A photographic essay of prolidase deficiency. Clin. Dysmorphol. 20(4):194–199. PMID: 21760498. doi: 10.1097/MCD.0b013e3283486cbd.
Freij, B.J., and der Kaloustian, V.M. 1986. Prolidase deficiency: A metabolic disorder presenting with dermatologic signs. Int. J. Dermatol. 25(7):431–443. PMID: 3771038. doi: 10.1111/j.1365-4362.1986.tb03446.x.
Goodman, S.I., Solomons, C.C., Muschenheim, F., McIntyre, C.A., Miles, B., and O’Brien, D. 1968. A syndrome resembling lathyrism associated with iminodipeptiduria. Am. J. Med. 45(1):152–159. PMID: 4968882. doi: 10.1016/0002-9343(68)90016-8.
Kelly, J.J., Freeman, A.F., Wang, H., Cowen, E.W., and Kong H.H. 2010. An Amish boy with recurrent ulcerations of the lower extremities, telangiectases of the hands, and chronic lung disease. J. Am. Acad. Dermatol. 62(6):1031–1034. PMID: 20466176. doi: 10.1016/j.jaad.2009.12.038.
Kurien, B.T., D’Sousa, A., Bruner, B.F., Gross, T., James, J.A., Targoff, I.N., Maier-Moore, J.S., Harley, I.T.W., Wang, H., and Scofield, R.H. 2013. Prolidase deficiency breaks tolerance to lupus-associated antigens. Int. J. Rheum. Dis. 16(6):674–680. PMID: 24330273. doi: 10.1111/1756-185X.12254.
Shrinath, M., Walter, J.H., Haeney, M., Couriel, J.M., Lewis, M.A., and Herrick A.L. 1997. Prolidase deficiency and systemic lupus erythematosus. Arch. Dis. Child. 76(5):441–444. PMID: 9196362. doi: 10.1136/adc.76.5.441.
Novel mutation in the FLG gene and its relationship with severe atopic dermatitis: a case report
Lucy Dong Xuan Lia, Julia Uptona
aDivision of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
Background: Cutaneous manifestations are common in primary immunodeficiencies (PIDs) affecting between 40% and 70% of patients (
Al-Herz and Nanda 2011). Aside from skin infections, high rates of atopic dermatitis have been reported in paediatric patients with PIDs with 13%–22% being affected by eczema (
Berron et al. 2000;
Al-Herz and Nanda 2011). In one study, 75 patients with severe atopic dermatitis underwent immune evaluation with 7.9% being diagnosed with an underlying PID (
Aghamohammadi 2014). There are various PIDs that are associated with severe atopic dermatitis. These include conditions such as but not limited to, autosomal dominant and recessive hyper-IgE syndrome, phosphoglucomutase 3 deficiency (PGM3), immune dysregulation, polyendocrinopathy, and enteropathy (IPEX), wiskott-aldrich syndrome (WAS), and CARD11 deficiency.
More often, atopic dermatitis is not associated with an underlying PID. Atopic dermatitis is a chronic inflammatory skin disease caused by a combination of various genetic and environmental factors that disrupts epidermal homeostasis. The epidermal barrier, particularly the stratum corneum, is the first line of defense between the host and the environment. Filaggrin (FLG), the major structural protein in the stratum corneum, plays a pivotal role in skin barrier function. As such, individuals with loss of function mutations in FLG are more prone to developing and having persistent severe atopic dermatitis with increased risks of skin infections, allergic sensitization, and asthma compared to patients without FLG mutations (
McAleer and Irvine 2013).
Palmer et al. (2006) showed that two independent loss of function genetic variants (R510X and 2282del4) in FLG were very strong predisposing factors for atopic dermatitis (
Palmer et al. 2006). The authors indicated that in their cohort of 15 families with ichthyosis vulgaris, atopic dermatitis was inherited in a semi-dominant trait with high penetrance in FLG-null homozygotes or compound heterozygotes and reduced penetrance in heterozygotes. A study in a Japanese cohort of 7 patients with ichthyosis vulgaris identified heterozygosity for 2 novel mutations within the
FLG gene which were subsequently found to be present in 8 out of 143 patients with atopic dermatitis. These same mutations were not found in 156 unrelated, nonatopic and nonichthyotic controls (p = 0.0015) (
Nomura 2007).
We report a paediatric patient with severe atopic dermatitis who was initially referred to Immunology for query PID. Genetic analysis from research whole exome sequencing has shown that the patient is heterozygous for a novel mutation in the FLG gene predicted to lead to a complete loss of function of FLG from that allele. We postulate that this mutation likely explains her clinical symptoms of atopy.
Methods: Clinical data was collected from retrospective chart review and directly from the patient and parents. Informed written consent was obtained from the parents. Blood samples were collected for functional and quantitative immune investigations. DNA was obtained and sent for clinical and research based genetic analysis.
Results:
Clinical Features: The patient is currently a 7 year old girl who was referred to Immunology for a query primary immunodeficiency in the context of severe atopic dermatitis that began at 5 months of age. She first presented to us at 3 years of age on Methotrexate therapy. From an infection standpoint, she had multiple MSSA skin infections all related to her underlying atopic dermatitis that resolved with appropriate courses of oral antibiotics. She had one admission secondary to staph scalded skin syndrome requiring IV antibiotics. There was no history of abscesses. There were no frequent sinopulmonary infections as a toddler. She had an episode of molluscum contagiosum around the genital area that did not require any treatment. She had no issues with dentition and did not have any other symptoms of autoimmunity. Her family history is significant for a maternal grandmother having severe atopic dermatitis and a paternal family history of autoimmunity including her father being HLAB27+ with reactive arthritis, a paternal uncle with vitiligo, and her paternal grandfather with ankylosing spondylitis.
In addition to her atopic dermatitis, the patient has multiple food allergies as well as a diagnosis of asthma for which she currently remains on Alvesco therapy.
Investigations: Laboratory investigations to date have shown appropriate lymphocyte immunophenotyping as well as immune function. Clinical genetic analysis has shown that she carries heterozygous variants of uncertain significance (VUS) in DOCK8, IL12RB1, and VPS13B. She was also found to be homozygous for a VUS in the NOD2 gene. Research whole exome sequencing has since revealed that she is heterozygous for a novel mutation in the FLG gene predicted to lead to complete loss of function from that allele.
Conclusions: We report a paediatric patient with a novel mutation in the FLG gene with severe atopic dermatitis, asthma, and multiple food allergies. While atopic dermatitis can be associated with various PIDs, defects in the FLG gene should be considered in patients with refractory atopic dermatitis. The identification of likely pathogenic mutations is important for better understanding of genotype-phenotype associations in patient populations with severe atopic dermatitis.
REFERENCES
Aghamohammadi, A., Moghaddam, Z.G., Abolhassani, H., Hallaji, Z., Mortazavi, H., Pourhamdi, S., Mohammadinejad, P., and Rezaei, N. 2014. Investigation of underlying primary immunodeficiencies in patients with severe atopic dermatitis. Allergol. Immunopathol. 42: 336–341. PMID: 23735167. doi: 10.1016/j.aller.2013.02.004.
Al-Herz, W., and Nanda, A. 2011. Skin manifestations in primary immunodeficient children. Pediatr. Dermatol. 28: 494–501. PMID: 21453308. doi: 10.1111/j.1525-1470.2011.01409.x.
Berron-Ruiz, A., Berron-Perez, R., and Ruiz-Maldonado, R. 2000. Cutaneous markers of primary immunodeficiency diseases in children. Pediatr. Dermatol. 17: 91–96. PMID: 10792794. doi: 10.1046/j.1525-1470.2000.01721.x.
McAleer, M.A., and Irvine, A.D. 2013. The multifunctional role of filaggrin in allergic skin disease. J. Allergy Clin. Immunol. 131: 280–291. PMID: 23374260. doi: 10.1016/j.jaci.2012.12.668.
Nomura, T., Sandilands, A., Akiyama, M., Liao, H., Evans, A.T., Sakai, K., Ota, M., Sugiura, H., Yamamoto, K., Sato, H., Palmer, C.N.A., Smith, F.J.D., McLean, W.H.I., and Shimizu, H. 2007. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J. Allergy Clin. Immunol. 119(2): 434–440. PMID: 17291859. doi: 10.1016/j.jaci.2006.12.646.
Palmer, C.N., Irvine, A.D., Terron-Kwiatkowski, A., Zhao, Y., Liao, H., Lee, S.P., Goudie, D.R., Sandilands, A., Campbell, L.E., Smith, F.J.D., O'Regan, G.M., Watson, R.M., Cecil, J.E., Bale, S.J., Compton, J.G., DiGiovanna, J.J., Fleckman, P., Lewis-Jones, S., Arseculeratne, G., Sergeant, A., Munro, C.S., Houate, B.E., McElreavey, K., Halkjaer, L.B., Bisgaard, H., Mukhopadhyay, S., and McLean, W.H.I. 2006. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 38(4): 441–446 PMID: 16550169. doi: 10.1038/ng1767.
Post-hematopoietic stem cell transplant infections in IKBKB deficiency
Lisa Lianga, Marianne Miguela, Geoff Cuvelierb, Tamar Rubina
aChildren’s Hospital Winnipeg, Division of Clinical Immunology and Allergy, Department of Pediatrics and Child Health, University of Manitoba, Winnipeg, Canada
bManitoba Blood and Marrow Transplant Program, CancerCare Manitoba, Division of Pediatric Hematology-Oncology-BMT, University of Manitoba, Winnipeg, Canada
Background: IKBKB deficiency is a rare, profound combined immunodeficiency involving functional activation defects in both innate and adaptive immunity (
Pannicke et al. 2013). A homozygous founder mutation in exon 13 of the
IKBKB gene (c.1292dupG) in the Northern Cree population in Manitoba is known to cause loss of IKKß expression, a critical component of the canonical IKK-nuclear factor kappa B (NF-kB) pathway. Affected individuals have presented in early infancy with life-threatening bacterial, viral, fungal and mycobacterial infections.
The largest IKBKB deficiency case series to date described 16 Northern Cree individuals with IKBKB deficiency, of whom 8 died of overwhelming infection prior to hematopoietic stem cell transplant (HSCT) (
Cuvelier et al. 2019). The other 8 patients underwent HSCT, although it is unknown if this treatment is curative for this condition. The 2 long-term survivors described in the series continued to develop new viral, bacterial, fungal and mycobacterial infections for many years post-HSCT despite lymphoid and myeloid donor chimerism, normal cell counts, normal T cell mitogen proliferative responses and normal immunoglobulin levels. In addition, those patients who underwent HSCT and received post-transplant vaccines failed to mount specific IgG. Of two patients diagnosed and transplanted pre-symptomatically because of a targeted newborn screening program in Manitoba (
Rubin et al. 2018), one passed away 11-months post-transplant, due to pneumococcal sepsis. The infection occurred while off of any anti-infective prophylaxis, although otherwise the standard laboratory parameters indicated excellent chimerism and full immune reconstitution. Thus, there was always a suspicion that that HSCT may not be fully curative of the immune deficiency in IKBKB deficiency.
At the time of publication of the case series, one of the 3 survivors was only 6 months post-HSCT, and longer term clinical outcome and immune reconstitution data was unknown. As well, since the case series was published, another patient was diagnosed and underwent HSCT for IKBKB deficiency, with additional post-HSCT infection-prevention precautions taken based on experience with prior patients. With anticipated introduction of province-wide newborn screening for IKBKB deficiency in Manitoba in the next few months, an appreciation of the long-term clinical and immune reconstitution outcomes for transplanted patients with IKBKB deficiency is essential.
Herein, we describe the post-HSCT clinical and immune outcomes of the 4 known surviving patients with homozygous c.1292dupG mutations in IKBKB.
Methods: A retrospective chart review was performed, and clinical features and immunologic data were extracted for all known surviving IKBKB deficient patients with homozygous c.1292dupG IKBKB mutations.
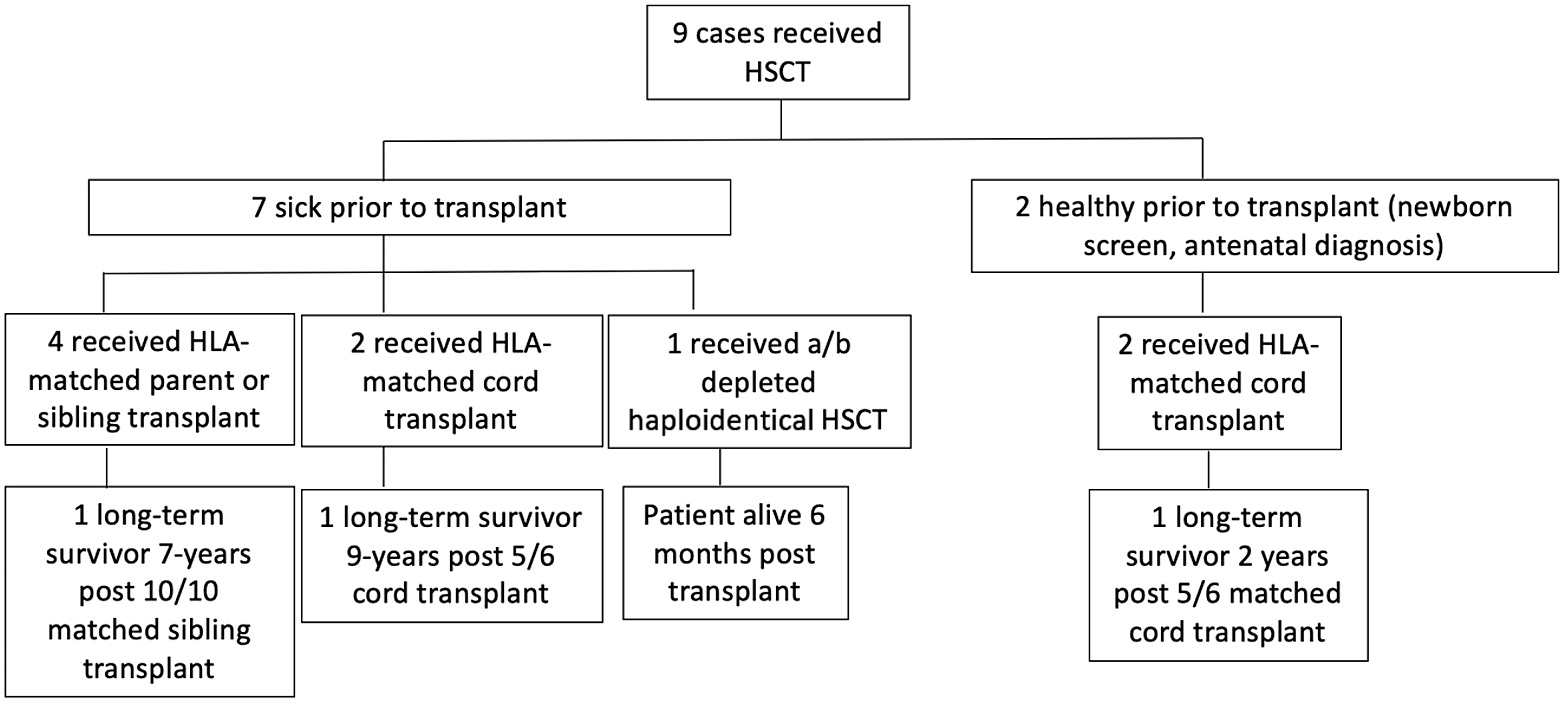
Results: There are 9 known cases of patients with homozygous c.1292dupG
IKBKB mutations who underwent HSCT in Manitoba. Of these, 7 were profoundly ill prior to HSCT, and 2 were diagnosed pre-symptomatically, through a targeted genetic newborn screening program (
Figure 1). Of the 9 patients who underwent HSCT, there are only 4 survivors (9 years, 7 years, 2 years and 6 months post-transplant).
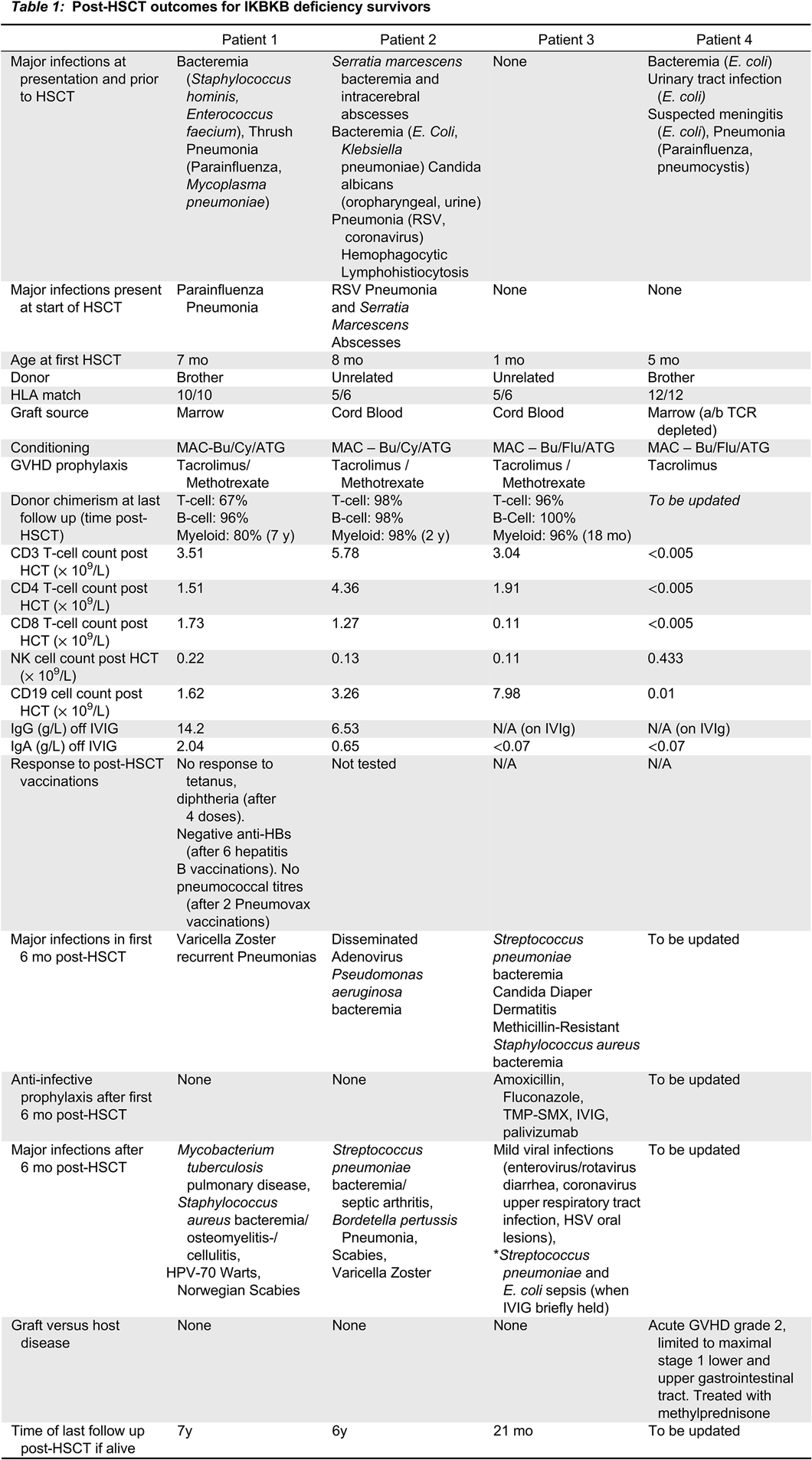
Patients 1 and 2 are long-term survivors (7 and 9 years respectively) and are described in a previous publication (
Cuvelier et al. 2019). Details regarding their post-HSCT prophylaxis, infections, and immune reconstitution are summarized in
Table 1.
Patient 3 was partially described in a previous publication (
Cuvelier et al. 2019). She was found to be homozygous for the founder loss of function IKBKB mutation after antenatal testing, performed because her older brother was diagnosed at birth through targeted genetic newborn screening for the founder mutation. Unfortunately, her older brother, also affected with IKBKB deficiency had died of
Streptococcus pneumoniae sepsis and meningitis 9 months post-HSCT, at 11 months old. At the time of death, he was not receiving IVIG or anti-pneumococcal prophylaxis.
Patient 3 was hospitalized immediately after birth and placed into isolation. She did not receive BCG or other vaccinations, and was started on IVIG, TMP-SMX, fluconazole and palivizumab prophylaxis. Starting at 1 month of age, she underwent myeloablative conditioning with busulfan, fludarabine and anti-thymocyeglobulin, and received a 5/6 matched unrelated cord graft. She experienced minor complications post-HSCT (mucositis, hypertension, TPN requirement and mild diarrhea), and never developed graft-versus-host disease (GVHD). Tacrolimus was discontinued by 5 months of age. Donor chimerism was >90% and she had early appearance of T cells at 3 months of age. However, she did also have marked unexplained lymphocytosis (primarily of B cells) from 6 to 18 months post-HSCT (B cells > 10 000 cell/mm3).
In the first 6 months post-HSCT, while on IVIG, amoxicillin, fluconazole and TMP-SMX (for Pneumocystis prophylaxis), patient 3 developed S. pneumoniae sepsis, central line associated S. aureus infection, recurrent S. aureus folliculitis, and chronic Candida diaper dermatitis. Between 6 and 18 months post-HSCT she developed generally mild viral illnesses (viral gastroenteritis, Coronavirus upper respiratory tract infection treated as an outpatient, Herpes simplex oral ulcers and chronic Candida dermatitis). Amoxicillin and fluconazole prophylaxis were continued, however, when IVIG was briefly held, as a trial, around 18 months post-HSCT, she developed S. pneumoniae and E. coli sepsis, and IVIG was resumed.
Finally, patient 4 was diagnosed at 4 months of age when he presented with Pneumocystis and Parainfluenza pneumonia, E. coli urinary tract infection and sepsis, and perianal malakoplakia. After complete treatment for his infections, he underwent a matched sibling donor bone marrow transplant following myeloablative conditioning. He developed grade 2 GVHD affecting his gastrointestinal tract, treated with a course of methylprednisolone. He had received GVHD prophylaxis with tacrolimus, and continued on prednisone post-discharge. He was also continued on anti-infective prophylaxis with amoxicillin, pentamadine, fluconazole, palivizumab, and IVIG, with a plan for the indefinite use of amoxicillin, IVIG and fluconazole.
Conclusions: IKBKB deficiency is a rare combined immune deficiency seen in the Northern Cree population in Manitoba. Of 17 known patients with the disease, there are only 4 current survivors. At greater than 6 months after HSCT, 3 of these 4 patients continued to develop significant and frequent infection, despite lymphoid and myeloid donor chimerism, normal cell counts, normal T cell mitogen proliferative responses and normal immunoglobulin levels. As well, the one surviving patient who received post-HSCT vaccinations was unable to mount normal responses to vaccination post-HSCT.
As with other defects involving the NFKB pathway, it is possible that HSCT does not correct the defect in cells of non-hematopoietic origin that are important in the immune response and also dependent on IKK/NFKB signaling. Immune reconstitution following transplant in IKBKB deficiency patients warrants further study, including detailed T cell studies pre- and post-HSCT, investigation of non-lymphoid cells (for example epithelial cells), and investigation of their secondary lymphoid organs such as the spleen and lymphatic tissue. However, for the time being, on the basis of the severe post-HSCT infections experienced in our cohort, we would suggest patients with IKBKB deficiency received indefinite anti-pneumococcal prophylaxis with both amoxicillin and immune globulin replacement, with consideration for ongoing prophylaxis against fungal infection.
REFERENCES
Cuvelier, G.D.E., Rubin, T.S., Junker, A., Sinha, R., Rosenberg, A.M., Wall, D.A., and Schroeder, M.L. 2019. Clinical presentation, immunologic features, and hematopoietic stem cell transplant outcomes for IKBKB immune deficiency. Clin. Immunol. 205:138–147. PMID: 30391351. doi: 10.1016/j.clim.2018.10.019.
Pannicke, U., Baumann, B., Fuchs, S., Henneke, P., Rensing-Ehl, A., RizzI, M., Janda, A., Hese, K., Schlesier, M., Holzmann, K., Borte, S., Laux, C., Rump, E.M., Rosenberg, A., Zelinski, T., Schrezenmeier, H., Wirth, T., Ehl, S., Schroeder, M.L., and Schwarz, K. 2013. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N. Engl. J. Med. 369(26):2504–2514. PMID: 24369075. doi: 10.1056/NEJMoa1309199.
Rubin, T.S., Rockman-Greenberg, C., Van Caeseele, P., Cuvelier, G.D.E., Kwan, L., and Schroeder, M.L. 2018. Newborn screening for IKBKB deficiency in Manitoba using genetic analysis. J. Clin. Immunol. 38(7): 742–744. PMID: 30288645. doi: 10.1007/s10875-018-0555-2.
A case of RAG1 mutation presenting with disseminated vaccine-strain induced varicella and vasculitis in a previously well infant
Mei Xua, Brenda Reida, Chaim M. Roifmana,b
aDivision of Clinical Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children and the University of Toronto, Toronto, ON, Canada
bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and the University of Toronto, Toronto, ON, Canada
Background: Recombination-activating gene 1 (
RAG1) and recombination-activating gene 2 (
RAG2) encode unique lymphocyte endonuclease proteins that are crucial in somatic V(D)J gene recombination (
Fugmann 2001;
Sadofsky 2001). T and B cell development is dependent on this process and there is a recognized spectrum of phenotypes which correlates with the level of recombination activity (
Delmonte et al. 2018;
Gennery 2019). We report a unique case of a patient with compound heterozygous
RAG1 variants, who was well until their initial presentation with vaccine-strain varicella.
Methods: Clinical data was gathered by retrospective chart review. Informed consent was obtained from the family.
Results:
Clinical Features: The patient was clinically well until she received her measles, mumps, rubella, and varicella vaccines at the age of 1. She subsequently developed fever, a vesicular rash and dusky purpura of the limbs. Family history revealed a sister who had passed away after presenting with a severe vasculitic rash as a child. Given the similarities, she was admitted and treated aggressively. A skin biopsy showed small vessel vasculopathy and vaccine-strain varicella was isolated from cerebral spinal fluid and skin lesions.
Initial Investigations: Her immune work-up was initially complicated by her illness, immunosuppressants, and IVIG. Following discontinuation of these, she continued to demonstrate low T and B-cell numbers, poor PHA, undetectable TRECS, abnormal expansion of TCR-Vbeta repertoire and impaired antibody responses. A thymic biopsy showed a lack of Hassall’s corpuscles with no clear corticomedullary distinction. Around the time of her initial presentation, genetic work-up with targeted sequencing was negative.
Management: Although there was no genetic diagnosis at the time, her laboratory findings of severe T-cell impairment, sibling’s course and thymic dysplasia were profound. After careful consideration, she proceeded to undergo a matched sibling donor bone marrow transplant (BMT) at the age of two. She had full engraftment with good evidence of immune reconstitution afterwards.
Post-Transplant investigations: Post-transplant, she had normal CBC and lymphocytes, normal immunoglobulins, protective vaccine titres, as well as normal numbers and functioning of T-cells and B-cells. Whole exome sequencing in 2014 identified a single missense mutation in RAG1, c.1420C>T. It wasn’t until recently, when whole genome sequencing revealed a second heterozygous missense variant, that her presentation could finally be explained.
Discussion and Conclusions: We present a previously well child who presented after her first live-viral vaccinations and was found to have severe T-cell dysfunction. Diagnostic confirmation of two
RAG1 missense variants was not found until several years after BMT, through whole genome sequencing. The c.1420C>T (p.Arg474Cys) mutation has previously been reported in cases of SCID, Omenn syndrome and CD4+ T cell lymphopenia (
Chen et al. 2014;
Kuijpers et al. 2011). Individuals with only this mutation have been noted to be healthy (
Chen et al. 2014). Therefore, the second variant is a novel mutation in exon 2 that has not been previously reported and leads to either complete or partial loss of function.
RAG1 gene defects lead to variable phenotypes, some of which are yet to be discovered (
Notarangelo et al. 2016). This case highlights that in the absence of genetic confirmation, clinical presentation, family history and immunological work-up are crucial in management decisions.
REFERENCES
Fugmann, S.D. 2001. RAG1 and RAG2 in V (D) J recombination and transposition. Immunol. Res. 23(1):23–40. PMID: 11417858. doi: 10.1385/IR:23:1:23.
Sadofsky, M.J. 2001. The RAG proteins in V (D) J recombination: More than just a nuclease. Nucleic Acids Res. 29(7):1399–1409. PMID: 11266539. doi: 10.1093/nar/29.7.1399.
Delmonte, O.M., Schuetz, C., and Notarangelo, L.D. 2018. RAG deficiency: Two genes, many diseases. J. Clin. Immunol. 38(6):646–655. PMID: 30046960. doi: 10.1007/s10875-018-0537-4.
Gennery, A. 2019. Recent advances in understanding RAG deficiencies. F1000Research 8.
Chen, K., Wu, W., Mathew, D., Zhang, Y., Browne, S.K., Rosen, L.B., McManus, M.P., Pulsipher, M.A., Yandell, M., Bohnsack, J.F., and Jorde, L.B. 2014. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations. J. Allergy Clin. Immunol. 133(3):880–882.e10. PMID: 24472623. doi: 10.1016/j.jaci.2013.11.038.
Kuijpers, T.W., IJspeert, H., van Leeuwen, E.M., Jansen, M.H., Hazenberg, M.D., Weijer, K.C., Van Lier, R.A., and van der Burg, M. 2011. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood. 117(22):5892–5896. PMID: 21502542. doi: 10.1182/blood-2011-01-329052.
Notarangelo, L.D., Kim, M.S., Walter, J.E., and Lee, Y.N. 2016. Human RAG mutations: Biochemistry and clinical implications. Nat. Rev. Immunol. 16(4):234–246. PMID: 26996199. doi: 10.1038/nri.2016.28.
Case report: delayed presentation of Ataxia-Telangiectasia
A. Bhattia, R. Bragera, A. Latchmanb, M. Kozenkjoc
aDepartment of Clinical Allergy and Immunology, McMaster University, Hamilton, ON, Canada
bDepartment of Pediatrics, McMaster University, Hamilton, ON, Canada
cDepartment of Genetics and Metabolics, McMaster University, Hamilton, ON, Canada
Background: We report a case of a child with a delayed diagnosis of Ataxia-Telangiectasia (AT) at 8 years of age. AT is a rare, autosomal recessive disorder, with a mean age of diagnosis of 2 years old. AT is associated with immunodeficiency, cerebellar neurodegenerative disease, and a predisposition to malignancy due to radiosensitivity and impaired T cell surveillance. AT patients may present in a variety of ways including ataxia, developmental delay, telangiectasias or ocular abnormalities. This case illustrates the importance of awareness and recognition of this disease amongst Primary Care and Pediatric practitioners.
Case Presentation: We report an 8 year-old male with a longstanding history of speech delay, followed remotely by Respirology for Interstitial Lung Disease, and more recently by Neurology for hypotonia and ataxia. He was referred to Genetics for further assessment and upon identification of the ATM gene change, was referred to our Immunology clinic. The patient had a history of one episode of mild community-acquired outpatient pneumonia and two episodes of acute otitis media. His growth curve followed the 5th %tile until the 7th year of life, where his weight and height dropped to < 3rd %tile. He had also been diagnosed with hypertrophic osteoarthropy of unknown significance, following admission for ongoing chest and lower limb bone pain. As part of the work up he had a CT chest which incidentally showed bilateral homogenous reticular shadows with subpleural sparing on CT. Subsequent bronchoalveolar lavage showed non-specific findings.
There was no family history of immune deficiency or malignancy; furthermore parents were not known to be consanguineous. Due to difficult social circumstances, including relocating to different cities, single parent family, and other challenges, there was a significant period where the patient was lost to follow up at age 6. The patient re-presented to care at age 8 years with worsening ataxia and abnormal eye movements. This led to genetic investigations and the pathogenic change in the ATM gene was identified on Whole Exome Sequencing (c.1290_1291delTG/c.7788+1G>T). Clinical evaluation at the time of work up revealed ataxia, failure to thrive, central hypotonia, and telangiectasias of the ear, sclera, and nose.
Laboratory analysis revealed serum alpha fetoprotein 190.8 ng/mL (normal <15.0). Initial immunoglobulins showed IgG 6.74, IgM 0.73, IgA< 0.07, and reduced T-cell numbers (CD3+ Absolute count 0.57, CD3+CD4+ Absolute count 0.36, CD3+CD8+ absolute count 0.18, CD19+ absolute count 0.14). Titres to tetanus toxoid and mumps demonstrated immunity, Rubella IgG was also reactive.
The patient was started on IVIG and Trimethoprim-Sulfamethoxazole prophylaxis and, after unsuccessful oral nutritional interventions, a G-tube was inserted. The family has been provided with increased social supports and has connected with local and international AT family support groups. The patient now has a comprehensive care team with close follow-up.
Conclusions: In summary, we report a case of delayed diagnosis of AT at 8 years of age following progressive speech delay, failure to thrive, ataxia and development of telangiectasia, in the absence of major clinical infection. This case demonstrates the importance of consideration of AT on the differential diagnosis of a child with a history of delayed motor milestones, interstitial findings on chest CT, and telangiectasias. Furthermore, these children are at increased risk of malignancy and should avoid radiation wherever possible, making early diagnosis imperative.
REFERENCES
Canman, C.E., and Lim, D.S. 1998. The role of ATM in DNA damage responses and cancer. Oncogene.
17(25): 3301–3308. PMID: 9916992. doi: 10.1038/
sj.onc.1202577.
Gatti, R., and Perlman, S. 1993–1999. Ataxia-telangiectasia. In GeneReviews. Edited by R.A. Pagon, T.D. Bird, C.R. Dolan, and K. Stephens. University of Washington, Seattle, WA.
Meijer, A.E., Zhivotovsky, B., and Lewensohn, R. 1999. Epstein-Barr virus-transformed lymphoblastoid cell lines of ataxia telangiectasia patients are defective in X-ray-induced apoptosis. Int. J. Radiat. Biol. 75(6): 709–716. PMID: 10405000. doi: 10.1080/095530099140041.
A case of failure to thrive, neutropenia, and rotavirus gastroenteritis in an infant
Meriem Latrousa, Catherine M. Biggsa,b, Stuart E. Turveya,b, Kyla J. Hildebranda,b
aDivision of Allergy and Immunology, Department of Paediatrics, British Columbia Children’s Hospital, The University of British Columbia, Vancouver, BC, Canada
bBritish Columbia Children’s Hospital Research Institute; Vancouver, BC, Canada
Background: Neutropenia, defined by an absolute neutrophil count (ANC) <1 × 10
9 cells/L in infants and <1.5 × 10
9 cells/L in older children, is not an uncommon finding in pediatric patients (
Angelino et al. 2013). Neutropenia can be categorized as acute if less than three months’ duration or chronic if 4 months or more (
Walkovich and Boxer 2013). The severity of neutropenia depends on the ANC: mild if between 1 and 1.5 × 10
9 cells/L, moderate if between 0.5 and 1 × 10
9 cells/L, and severe when below 0.5 × 10
9 cells/L (for patients older than 1 year of age). Frequency and severity of infections is known to be related to the degree of neutropenia. However, many patients with moderate–severe neutropenia remain well, suggesting that there are other factors influencing predisposition to infection, such as the speed of onset and the duration of neutropenia, bone marrow myeloid reserves, absolute monocyte count, and function of phagocytes (
Angelino et al. 2013).
There are a number of causes of neutropenia early in life caused by several inherited and acquired conditions including nutritional deficiencies, bone marrow failure syndromes, suppression in the setting of illness, and disorders of immune function. Intrinsic disorders of proliferation or maturation of myeloid precursor cells which may present with neutropenia include: primary disorders of myelopoiesis (cyclic neutropenia, severe congenital neutropenia, Kostmann syndrome), disorders of molecular processing (Schwachman-Diamond syndrome, dyskeratosis congenita), disorders of vesicular trafficking (Chédiak-Higashi syndrome, Griscelli syndrome type II, Cohen syndrome, Hermansky-Pudlak syndrome type II, p14 deficiency, VPS45 defects), disorders of metabolism (Glycogen storage disease type 1b, Methylmalonic/propionic acidemias, Glucose-6-phosphate catalytic subunit 3 deficiency, Barth syndrome, Pearson syndrome), and neutropenia in disorders of immune function (common variable immunodeficiency, IgA deficiency, severe combined immunodeficiency, Hyper-IgM syndrome, Warts, Hypogammaglobulinemia, Infections, and Myelokathexis syndrome (WHIM), Cartilage-hair hypoplasia, Schimke immunoosseous dysplasia, X-linked agammaglobulinemia) (
Kliegman et al. 2011). A large number of cases of neutropenia remains of unknown etiology, however (
Angelino et al. 2013).
Case: A term infant female was born via spontaneous vaginal delivery to a 35-year-old G1, P0 female who had a healthy pregnancy, normal prenatal screening tests, and normal prenatal ultrasounds. Birthweight was low at 2.215 kg (0.86 percentile). The patient was kept in hospital for 36 h for an initial low blood glucose and temperature instability which resolved spontaneously. On the 5th day of life, the family presented to the emergency department for jaundice. It was noted at that time that she had a low neutrophil count of 1.29 × 109 cells/L (normal range for age: 5 – 21 × 109 cells/L). Her lymphocyte count was normal at 6.01 × 109 cells/L.
At 3–4 weeks of age, she developed oral thrush that persisted over time, however, was not treated with Nystatin oral suspension until 9 weeks of age. Shortly after starting this treatment, she began to have 6–8 non-bloody, green-yellow loose stools per day, with no associated fever. The next day, she received her 2-month immunizations including the Diphtheria, Tetanus, Pertussis, Hepatitis B, Haemophilus influenzae type b, Meningococcal C Conjugate, Pneumococcal Conjugate and Rotavirus vaccines whereby non-bloody loose stools progressed and worsened over the next few days. She experienced an elevated temperature (parental report) on the day of the vaccines which resolved. She was brought to hospital at 10 weeks of life due to low oral intake and was admitted under the General Pediatrics team. The Immunology service was consulted 2 days later for rotavirus positive stool, skin rash, oral thrush, failure to thrive, and neutropenia.
The infant had no history of infection. Seborrheic dermatitis was present since birth. A dry skin rash began at approximately 3 weeks of age and had been managed at home with topical application of coconut oil. She had had no neurological symptoms and there were no concerns with regard to her development. She had no history of surgeries or hospitalizations otherwise. There were no known allergies, and she took no medications on a daily basis. Her weight had been closely monitored by her pediatrician.
The parents are of a non-consanguineous union. Parents are of African Indian descent and Chinese descent. She is the only child born to her parents, and there are no previous pregnancies in her mother. The mother was reported to be healthy with no health issues. Her brother’s wife has a history of two previous spontaneous abortions. Maternal uncle died of pancreatic cancer. The father, fathers’ two siblings, and maternal and paternal grandparents were healthy. There is a history of colon cancer and breast cancer in the maternal great grandmothers. No family history of autoimmunity, primary immune deficiency, neutropenia, blood disorders or malignancies, and no history of sudden death among infants or stillbirths.
Physical examination revealed a non-dysmorphic infant female who appeared small for age. Vitals were within normal limits. Growth parameters on admission showed a weight of 3.43 kg, length of 50.5 cm, and head circumference of 35.5 cm, all well below the 3rd percentile on World Health Organization growth curves. Oral thrush was noted on the tongue. Her skin was dry and eczematous in appearance particularly on the face and trunk, with underlying erythema diffusely. No diaper dermatitis. Nails and hair were healthy with no dystrophy. Chest, heart, abdominal exams were normal, with no organomegaly. Small lymph nodes were palpated in the axilla and inguinal canal.
Initial investigations revealed a white blood cell count of 8.4 × 109 cells/L, neutrophils 0.94 × 109 cells/L (low, normal range 1–9 × 109 cells/L), lymphocytes 7.01 × 109 cells/L, monocytes 0.43 × 109 cells/L (low), eosinophils 0.01 × 109 cells/L (low). Hemoglobin was 118 g/L, platelets 452 × 109 cells/L. Subsequent flow cytometry of T and B subsets include absolute counts (× 109 cells/L) as follows: CD3 3.38, CD4 2.39, CD8 1.06, CD19 0.65, NK cells 0.35 (all normal for age). IgG level 3.4 g/L, IgA 0.07 g/L, IgM 0.38 g/L (all normal). IgE level was elevated at 26 g/L. Albumin 34 g/L, ALT 154 U/L (elevated), AST 250 U/L (elevated), ALP 379 U/L (elevated). Urinalysis was negative for protein. Stool sample was positive for rotavirus, negative for other viruses and bacteria. Thymus was present on chest x-ray.
Given her presentation of failure to thrive, oral thrush, and eczematous rash, severe combined immunodeficiency (SCID) was considered, as well as class switch recombination defects, reticular dysgenesis, WHIM, congenital neutropenia or a bone marrow failure syndrome such as Shwachman-Diamond Syndrome (SDS). Upon reassessment the day following initiation of topical steroids, the skin rash had nearly resolved with no underlying erythema remaining. She had improvement in her diarrhea and oral thrush. The patient was discharged from hospital and followed in the outpatient setting, with pending TCRV-beta studies and repeat CBC showing resolved neutropenia at 1.87 × 109 cells/L. Her liver enzymes however remained elevated.
The patient continued to do well at home, with resolved loose stool and improved eczematous rash and oral thrush. Her weight had been slow to gain back, and she was started on formula supplementation. She had otherwise been well with no fevers or concerns for new infections. Repeat investigations 3 weeks following discharge from hospital revealed a normal white blood cell count of 8.2 × 109 cells/L, however neutrophils were low again at 0.24 × 109 cells/L. Hemoglobin was 154 g/L, and platelets 405 × 109 cells/L. Flow cytometry revealed a diverse repertoire of T-cell receptors. Her liver enzymes, although improved, remained elevated. An abdominal ultrasound was normal. She was seen by the Gastroenterology and Hematology services due to slow bone marrow recovery, history of elevated fat content in the stool, poor weight gain and transaminitis. A bone marrow failure disorder was strongly suspected and molecular testing for SDS was sent. A skeletal survey showed no evidence of skeletal dysplasia. Fecal elastase was low at 160 μg/g stool, indicating a mild-to-moderate exocrine pancreatic insufficiency and increasing suspicion for a diagnosis of SDS.
At 5 months of age, a Blueprint Genetics Bone Marrow Failure Syndrome Panel revealed a Shwachman-Bodian-Diamond Syndrome (SBDS) heterozygous pathogenic variant c.258+2T>C and SBDS heterozygous pathogenic variant c.183_184delinsCT, p.(Lys62*) consistent with compound heterozygosity and SDS. Both pathogenic variants have been previously identified in other patients affected with SDS. Subsequent testing confirmed that each parent was a carrier of one of the pathogenic variants in the SBDS gene, in keeping with diagnosis of SDS.
Discussion: We present a case of an infant female with clinical features suspicious for inborn error of immunity including SCID, diagnosed with SDS on genetic testing. SDS is an inherited autosomal recessive bone marrow failure syndrome characterized by exocrine pancreatic dysfunction, short stature, skeletal abnormalities and neutropenia. Patients with SDS are at higher risk for progressive marrow failure, myelodysplastic syndrome (MDS), and acute myeloid leukemia (AML) (
Myers et al. 2014;
Nelson and Myers 2018). Presenting symptoms usually include steatorrhea and failure to thrive due to malabsorption, however symptoms may be subtle in some patients (
Kliegman et al. 2011).
Neutropenia is seen in nearly 70% of patients with SDS at the time of presentation and almost all patients on follow-up. It can be persistent or intermittent, and of varying degrees of severity. Neutrophils may have defects in mobility, migration, and chemotaxis due to alterations in neutrophil cytoskeletal or microtubular function (
Kliegman et al. 2011). Patients with SDS have also been found to have an increased tendency to recurrent bacterial, viral, and fungal infections beyond that attributable to neutropenia. Sepsis, for example, is one of the most common infections in patients with SDS leading to mortality (
Myers et al. 2013). In 2001, Dror et al. prospectively studied immune function in SDS patients. B-cell defects (low levels of IgG and IgG subclasses, low levels of circulating B cells, deficient antibody production, and decreased in vitro B lymphocyte proliferation) and T-cell defects (low circulating T lymphocytes or CD3/CD4 cell subpopulations or decreased in vitro T-lymphocyte proliferation) were described in most patients studied. Abnormal neutrophil chemotaxis was also reported, however compared to other disorders of neutrophil chemotaxis, patients with SDS preserve the ability to form purulent abscesses and empyema (
Dror et al. 2001).
The clinical diagnosis of SDS relies on evidence of bone marrow dysfunction as well as exocrine pancreatic insufficiency (
Kliegman et al. 2011). Differentiating SDS from dyskeratosis congenita may not be possible based only on clinical features, therefore telomere length measurement may help in distinguishing both entities. Approximately 90% of patients with SDS have pathogenic variants in the
SBDS gene on chromosome 7q11, which encodes a protein involved in ribosomal maturation and is implicated in acute myeloid leukemia (AML) (
Kliegman et al. 2011;
Myers et al. 2014). Additional variants have recently been discovered in genes such as DnaJ Heat Shock Protein Family (Hsp40) Member C21 (
DNAJC21)
, Elongation Factor Like GTPase 1 (
EFL1)
, and Signal Recognition Particle 54 (
SRP54). The timely diagnosis of SDS carries many implications for medical management (Myers et al. 2014). Daily administration of subcutaneous G-CSF has successfully stimulated a sustained increase in neutrophil counts in SDS patients (
Kliegman et al. 2011). Routine monitoring of blood counts and bone marrow cytogenetics allow for early recognition of bone marrow failure, myelodysplastic syndrome, and acute myeloid leukemia. Hematopoietic stem cell transplant (HSCT) is the only curative option for severe marrow failure, myelodysplastic syndrome, and leukemia. Patients with SDS require reduced intensity conditioning regimens to avoid drug-related toxicity, thus, identification of the underlying diagnosis of SDS is critical (
Kliegman et al. 2011;
Myers et al. 2014).
Conclusions: Although it is imperative to rule-out SCID in the context of failure to thrive and persistent diarrhea following rotavirus immunization, SDS and other bone marrow failure disorders should be considered on the differential diagnosis of patients presenting with neutropenia, diarrhea, and failure to thrive in infancy. Early recognition and diagnosis of SDS carries implications for symptom management, prevention of associated morbidity and mortality, and choice of conditioning regimens for HSCT.
REFERENCES
Angelino, G., Caruso, R., D'Argenio, P., Carducci, F.I.C., Pascone, R., Lanciotti, M., Cancrini, C., Palma, P., Aiuti, A., Rossi, P., and Finocchi, A. 2013. Etiology, clinical outcome, and laboratory features in children with neutropenia: Analysis of 104 cases. Pediatr. Allergy Immunol. 25(3):283–289. PMID: 24325465. doi: 10.1111/pai.12177.
Dror, Y., Ginzberg, H., Dalal, I., Cherepanov, V., Downey, G., Durie, P., Roifman, C.M., and Freedman, M.H. 2001. Immune function in patients with Shwachman–Diamond syndrome. Br. J. Hematol. 114(3):712–7. doi: 10.1046/j.1365-2141.2001.02996.x.
Kliegman, R.M., Stanton, B.F., St. Geme III, J.W., Schor, N.F., and Behrman, R.E. 2011. Nelson textbook of pediatrics. 19th ed. Elsevier Saunders, Philadelphia, PA.
Myers, K., Davies, S., and Shimamura, A. 2013. Clinical and molecular pathophysiology of Shwachman–Diamond syndrome: An update. Hematol. Oncol. Clin. North Am. 27(1):117–128. PMID: 23351992. doi: 1016/j.hoc.2012.10.003.
Myers, K., Bolyard, A.A., Otto, B., Wong, T.E., Jones, A.T., Harris, R.E., Davies, S.M., Dale, D.C., and Shimamura, A. 2014. Variable clinical presentation of Shwachman–Diamond syndrome: Update from the North American Shwachman–Diamond Syndrome Registry. J. Pediatr. 164(4):866–870. PMID: 24388329. doi: 10.1016/j.jpeds.2013.11.039.
Nelson, A.S., and Myers, K.C. 2018. Diagnosis, treatment, and molecular pathology of shwachman-diamond syndrome. Hematol. Oncol. Clin. North Am. 32(4):687. PMID: 30047420.
Walkovich, K., and Boxer, L.A. 2013. How to approach neutropenia in childhood. Pediatr. Rev. 34(4):173–184. PMID: 23547064. doi: 10.1542/pir.34-4-173.
Not just SCID: a cohort of non-SCID newborn screen positive infants with low TRECs and lymphopenia
Yehonatan Pasternaka, Amarilla Mandolaa, Ori Scotta, Brenda Reida, Chaim M. Roifmana,b
aDivision of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
bCanadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and Research Institute, Toronto, ON, Canada
Introduction: Neonatal T-cell receptor excision circles (TRECs) screening can allow pre-symptomatic diagnosis of Severe Combined Immune Deficiency (SCID) leading to early diagnosis and treatment (
Chan and Puck 2005). TRECs are short segments of DNA excised from the T-cell receptor upon maturation of developing T cells in the thymus. Therefore, TRECs are a marker for recently formed T-lymphocytes, while conversely, absence or strongly reduced levels of TRECs indicates a low level of newly formed T-lymphocytes (
van der Burg et al. 2019).
This test, while very sensitive for detecting typical SCID and most hypomorphic forms of SCID, is not specific, and only some of the children examined for low T cells will in fact have SCID (
Kwan et al. 2014;
Amatuni et al. 2019) Other causes of low TRECs can be divided into other primary immunodeficiencies (PIDs) or syndromes related to immunodeficiencies, and for other reasons for low T-cells such as prematurity, maternal illness, or the prenatal use of immunosuppressive or immunomodulatory medications (
Patrawala and Kobrynski 2019). With the wide implementation of Next Generation Sequencing (NGS) in the evaluation of PID, more cases of non-SCID infants with immune deficiency and positive newborn screen (NBS) with low TRECs are having genetic diagnoses.
In 2013, the Ontario Ministry of Health added the TREC assay for SCID to the uniform NBS panel (
Newborn Screening Ontario 2020). A specific algorithm was developed for assessing and referring NBS positive cases (
Reid et al. 2017). We herein describe our experience with non-SCID NBS positive infants with low TRECs and ongoing lymphopenia who were referred to our center through the Ontario NBS program between the years 2013–2020.
Methods: We performed a chart review of patients diagnosed with low TRECs on NBS, referred to our center through the Ontario NBS program. Cut-off TREC levels were defined as 75 TRECs/µL. SCID patients were defined according to PIDTC criteria (
Shearer et al. 2014) Clinical, demographic, and laboratory data for patients who had repeated low TRECs and/or T cell lymphopenia in the cohort were analyzed. Written informed consent was obtained from the parents or guardians of the participants of this study.
Results: 151 infants with low TRECs levels (<75 TRECs/µL) on initial NBS were referred to our immunology service since the introduction of TREC into the NBS. 114 (75.5%) patients had normal TRECs levels on repeated testing or died before the assay could be repeated. The cases in this group were associated with prematurity, maternal immunosuppression and corticosteroids treatment, and post-cardiac thymectomy. Out of the 37 patients with repeated low TRECs and/or lymphopenia, 14 patients (38%) were diagnosed as having SCID.
23 non-SCID patients diagnosed through NBS had repeated low TRECs or ongoing lymphopenia. For breakdown of patients’ diagnoses see
Figure 1. 16 of the cases (69.5%) were male, median age was 3.6 years. All patients are currently alive. The largest sub-group of this cohort of cases was patients with syndromes associated with immunodeficiency (ID, e.g. Jacobsen syndrome). The most common single diagnoses were Ataxia Telangiectasia (AT) and
FOXN1 heterozygous mutations followed by DiGeorge syndrome. Demographic, clinical and laboratory characteristics of non-SCID NBS positive infants with low TRECs are presented in
Table 1.
Discussion: Most cases of low TRECs on NBS are due to etiologies other than ID, most commonly prematurity. However, those patients tend to recover their T-cell lymphopenia. In our cohort, none of the living patients who had normalized their initial low TRECs levels without lymphopenia were diagnosed with ID. Our study focused on the non-SCID patients detected by NBS, presenting with ongoing T-cell lymphopenia. The cohort demonstrates high yield of laboratory and genetic testing in those patients, with high detection rate of PID and syndromes associated with ID. Only 17% of patients with positive NBS and lymphopenia did not have a diagnosis, most of them under the age of 2 years.
The most common single diagnoses in our study were AT and
FOXN1 heterozygous mutations. AT is an autosomal recessive syndrome encompassing progressive neuronal degeneration, ocular and cutaneous telangiectasias, variable immunodeficiency, and cancer susceptibility. The relatively high frequency of AT in our cohort suggests that this diagnosis should be considered in cases with NBS positivity. Those patients were presented in detail in a study by
Mandola et al. (2019). As clinical presentation of AT tend to be variable, patients who were diagnosed through NBS benefits from early medical interventions and multidisciplinary follow up.
FOXN1 is the master regulatory gene of thymic epithelium development. Recently, heterozygous
FOXN1 mutations have been identified as a genetic etiology of newborn T-cell lymphopenia (
Bosticardo et al. 2019). The cases of
FOXN1 in our cohort were characterized by mild to moderate T-cell lymphopenia which improved over time and were presented in detail in a study by Scott et al. (in print).
A recent study by Maraucher et al., evaluating 100 articles describing outcomes of positive NBS cases found that the overall most frequently recorded PID was DiGeorge syndrome (most frequently 22q11.2 deletion syndrome) (
Mauracher et al. 2017). Our study is in keeping with this previous report. However, many patients with DiGeorge are relatively easily detected in the newborn period due to additional findings such as cardiac anomalies and hypocalcemia.
The data reported herein, although representing a catchment area of 10 million people, describes the experience of a single center. When comparing our results to those presented by Mauracher et al. in their extensive literature search, the portion of PID patients in our cohort is somewhat lower (22% vs. 40%). This might represent a publication bias as positive cases are more often published. Importantly, our data also stresses the importance of T-cell lymphopenia as a predictor of significant diagnosis.
Low TRECs at birth need to be taken seriously, because they have been associated with an increased mortality, even when excluding SCID patients (
Tierce et al. 2013). Although the diagnostic algorithm for infants with suspected SCID is well described (
Van Der Spek et al. 2015), 10 years after the institution of SCID NBS, there is no unified definition of T-Cell Lymphopenia, no standard diagnostic algorithm, and no clinical care pathways for non-SCID patients diagnosed through NBS (
Patrawala and Kobrynski 2019). At our center, patients with positive NBS and lymphopenia are followed closely and, as presented in our cohort, often receive definitive diagnosis. This emphasizes the need for extensive clinical, laboratory and genetic work up in the care of this population.
Conclusion: Patients with low TRECs on NBS and ongoing lymphopenia are likely to have a PID or a syndrome associated with PID. Close monitoring of this population, together with extensive investigation leads to early detection of various immunodeficiencies, and therefore for better care and management.
REFERENCES
Amatuni, G.S., Currier, R.J., Church, J.A., Bishop, T., Grimbacher, E., Nguyen, A.A., Agarwal-Hashmi, R., Aznar, C.P., Butte, M.J., Cowan, M.J., and Dorsey, M.J. 2019. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California, 2010–2017. Pediatrics. 143(2):e20182300. PMID: 30683812. doi: 10.1542/peds.2018-2300.
Bosticardo, M., Yamazaki, Y., Cowan, J., Giardino, G., Corsino, C., Scalia, G., Prencipe, R., Ruffner, M., Hill, D.A., Sakovich, I., and Yemialyanava, I. 2019. Heterozygous FOXN1 variants cause low TRECs and severe T cell Lymphopenia, revealing a crucial role of FOXN1 in supporting early Thymopoiesis. Am. J. Hum. Genet. 105(3):549–561. PMID: 31447097. doi: 10.1016/j.ajhg.2019.07.014.
Chan, K., and Puck, J.M. 2005. Development of population-based newborn screening for severe combined immunodeficiency. J. Allergy Clin. Immunol. 115(2):391–398. PMID: 15696101. doi: 10.1016/j.jaci.2004.10.012.
Kwan, A., Abraham, R.S., Currier, R., Brower, A., Andruszewski, K., Abbott, J.K., Baker, M., Ballow, M., Bartoshesky, L.E., Bonagura, V.R., and Bonilla, F.A. 2014. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 312(7):729–738. PMID: 25138334. doi: 10.1001/jama.2014.9132.
Mauracher, A.A., Pagliarulo, F., Faes, L., Vavassori, S., Güngör, T., Bachmann, L.M., and Schmid, J.P. 2017. Causes of low neonatal T-cell receptor excision circles: A systematic review. J. Allergy Clin. Immunol. 5(5):1457–1460. PMID: 28359806. doi: 10.1016/j.jaip.2017.02.009.
Patrawala, M., and Kobrynski, L. 2019. Nonsevere combined immunodeficiency T-cell lymphopenia identified through newborn screening. Curr. Opin. Allergy Clin. Immunol. 19(6):586–593. PMID: 31490207. doi: 10.1097/ACI.0000000000000586.
Reid, B., Ovadia, A., and Schejter, Y.D. 2017. Managing newborn screening for SCID in a referral centre. LymphoSign J. 4(2):77–9.
Mandola, A.B., Reid, B., Sirror, R., Brager, R., Dent, P., Chakroborty, P., Bulman, D.E., and Roifman, C.M. 2019. Ataxia telangiectasia diagnosed on newborn screening–Case cohort of 5 years’ experience. Front. Immunol. 10:2940. PMID: 31921190. doi: 10.3389/fimmu.2019.02940.
Scott, O., Mandolla, A., Pasternak, Y., Dinur-Schejter, Y., Reid, B., Hong-Diep Kim, V., and Roifman, C. in print. Heterozygous variants in FOXN1 among patients with abnormal T-cell receptor excision circles and lymphopenia: A single-centre experience. LymphoSign J.
Shearer, W.T., Dunn, E., Notarangelo, L.D., Dvorak, C.C., Puck, J.M., Logan, B.R., Griffith, L.M., Kohn, D.B., O’Reilly, R.J., Fleisher, T.A., and Pai, S.Y. 2014. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: The Primary Immune Deficiency Treatment Consortium experience. J. Allergy Clin. Immunol. 133(4):1092–1098. PMID: 24290292. doi: 10.1016/j.jaci.2013.09.044.
Tierce, M.L., Ruehle, M., Andruszewski, K., Cavanagh, K., Dahl, K., Kleyn, M., Long, P.M., Walkovich, K., Wood, H., Young, W., and Secord, E.A. 2013. TREC <30 in infants without SCID as a marker for high mortality. J. Allergy Clin. Immunol. 131(2):AB73. doi: 10.1016/j.jaci.2012.12.925.
Van der Burg, M., Mahlaoui, N., Gaspar, H.B., and Pai, S.Y. 2019. Universal newborn screening for severe combined immunodeficiency (SCID). Front. Pediatr. 7:373. PMID: 31620409. doi: 10.3389/fped.2019.00373.
Van Der Spek, J., Groenwold, R.H., Van Der Burg, M., and van Montfrans, J.M. 2015. TREC based newborn screening for severe combined immunodeficiency disease: A systematic review. J. Clin. Immunol. 35(4):416–430. PMID: 25893636. doi: 10.1007/s10875-015-0152-6.
Non-infectious cystitis in an adult with STAT3 gain-of-function mutation
Erika Yue Leea,b, Julia Uptona,b,c, Michelle Sholzberga,d, Luke Reynoldse, Rola Saleeba,f, Dory Abosha,d, Stephen Betschela,b
aDepartment of Medicine, University of Toronto, Toronto, ON, Canada
bDivision of Clinical Immunology and Allergy, Department of Medicine, St. Michael’s Hospital, Toronto, ON, Canada
cClinical Immunology and Allergy, Department of Pediatrics, Hospital for Sick Children, Toronto, ON, Canada
dDivision of Hematology, St. Michael’s Hospital, Toronto, ON, Canada
eSection of Surgery, Department of Urology, University of Chicago, Chicago, IL, USA
fDepartment of Pathology, St. Michael’s Hospital, Toronto, ON, Canada
Background: Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that plays a critical role in regulating cellular proliferation, survival and differentiation (
Flanagan et al. 2014;
Forbes et al. 2016). Mutations in STAT3 resulting in either loss-of-function (LOF) or gain-of-function (GOF) have been described (
Forbes et al. 2016). While STAT3 LOF results in hyper-IgE syndrome, STAT3 GOF is linked to autoimmunity and lymphoproliferation (
Forbes et al. 2016). The latter is inherited in an autosomal dominant manner, and mutations can be either germline or de novo (
Flanagan et al. 2014;
Fabre et al. 2019). STAT3 GOF was first described in 2014 (
Flanagan et al., 2014) and various mutations have been reported (
Fabre et al. 2019) (
Figure 1). It is characterized by short stature and early-onset multi-organ dysfunction, including those due to autoimmunity, lymphoproliferation and recurrent infections (
Fabre et al. 2019).
Herein, we describe a 26-year-old male who initially presented with recurrent sinopulmonary infections since 6 years of age, requiring frequent oral antibiotics. He also developed neutropenia, lymphopenia, immune thrombocytopenia (ITP) and splenomegaly. At 19 years of age, he was admitted to the hospital with febrile neutropenia due to pneumonia. While his immunoglobulins were always within normal limits, he had IgG2 and 4 subclass deficiency, and failed to mount an adequate vaccine response to pneumococcal antigen. He was thus diagnosed with symptomatic dysgammaglobulinemia and started on immunoglobulin replacement. At 23 years of age, he developed warm autoimmune hemolytic anemia and neutropenia. Because of his lack of response to prednisone, he was given 4 doses of rituximab and recovered. At 26 years of age, he developed frequent urination and dysuria. Urine culture showed no bacterial growth and initial pelvic ultrasound showed circumferential bladder thickening.
His other past medical history is notable for history of pulmonary tuberculosis infection (treated), uncomplicated EBV infection and G6PD deficiency. He was born in Philippines and moved to Canada at the age of 14. His family history was non-contributory.
Methods: Genetic testing through a commercial PID panel was sent. Thorough work-up for bladder wall inflammation was done; it included imaging, biopsy and microbiological cultures.
Results: Patient was diagnosed with STAT GOF mutation. His genetic testing showed heterozygous mutation for
STAT3 c.2144C>T, p.(Pro715Leu), which is pathogenic. The location of the mutation in the gene is shown in
Figure 1. Further, the initial CT urogram showed diffuse circumferential bladder wall inflammation and bilateral hydroureteronephrosis (
Figure 2). Patient underwent two biopsies of the bladder wall (percutaneous biopsy then transurethral biopsy), which were negative for malignancy but showed chronic inflammatory changes. Thorough infectious work-up was all negative. It included bacterial, fungal, and mycobacterial cultures for his bladder biopsies as well as for three consecutive samples of urine and stool, three consecutive stool cultures for ova and parasite, urine PCR for ureaplasma, and serology for schistosomiasis.
Conclusion: We present a patient with early-onset autoimmune cytopenia, lymphadenopathy, splenomegaly and dysgammaglobulinemia due to STAT3 GOF mutation. He developed bladder wall inflammation causing bilateral hydrouretero-nephrosis and renal dysfunction; he was treated with prednisone that led to a complete clinical as well as biochemical but partial radiographic response. While the treatment for bladder wall inflammation due to lymphoproliferation is unclear, the use of immunosuppressants such as prednisolone and tacrolimus for non-infectious interstitial cystitis has been reported (
Kaneko et al. 2012). On the other hand, Jak inhibitors such as ruxolitinib or tofacitinib for patients with more severe systemic involvement due to STAT3 GOF has also been reported (
Forbes et al. 2018). While the same mutation found in our patient has been described in four other patients in the literature (Forbes et al. 2018), to the best of our knowledge, bladder wall inflammation in the context of STAT3 GOF has not been reported. Our case illustrates a possibly new albeit atypical presentation of this rare genetic condition known to cause lymphoproliferation.
REFERENCES
Fabre, A., Marchal, S., Barlogis, V., Mari, B., Barbry, P., Rohrlich, P.S., Forbes, L.R., Vogel, T.P., and Giovannini-Chami, L. 2019. Clinical aspects of STAT3 gain-of-function germline mutations: A systematic review. J. Allergy Clin. Immunol. Pract. 7(6):1958–1969.e9. PMID: 30825606. doi: 10.1016/j.jaip.2019.02.018.
Flanagan, S.E., Haapaniemi, E., Russell, M.A., Caswell, R., Allen, H.L., De Franco, E., McDonald, T.J., Rajala, H., Ramelius, A., Barton, J., Heiskanen, K., Heiskanen-Kosma, T., Kajosaari, M., Murphy, N.P., Milenkovic, T., Seppänen, M., Lernmark, Å., Mustjoki, S., Otonkoski, T., Kere, J., Morgan, N.G., Ellard, S., and Hattersley, A.T. 2014. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat. Genet. 46(8):812–814. PMID: 25038750. doi: 10.1038/ng.3040.
Forbes, L.R., Milner, J., and Haddad, E. 2016. Signal transducer and activator of transcription 3: A year in review. Curr. Opin. Hematol. 23(1):23–27. PMID: 26574998. doi: 10.1097/MOH.0000000000000206.
Forbes, L.R., Vogel, T.P., Cooper, M.A., Castro-Wagner, J., Schussler, E., Weinacht, K.G., Plant, A.S., Su, H.C., Allenspach, E.J., Slatter, M., Abinun, M., Lilic, D., Cunningham-Rundles, C., Eckstein, O., Olbrich, P., Guillerman, R.P., Patel, N.C., Demirdag, Y.Y., Zerbe, C., Freeman, A.F., Holland, S.M., Szabolcs, P., Gennery, A., Torgerson, T.R., Milner, J.D., and Leiding, J.W. 2018. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J. Allergy Clin. Immunol. 142(5):1665–1669. PMID: 30092289. doi: 10.1016/j.jaci.2018.07.020.
Kaneko, G., Nishimoto, K., Ito, Y., and Uchida, A. 2012. The combination therapy of prednisolone and tacrolimus for severe painful bladder syndrome/interstitial cystitis. Can. Urol. Assoc. J. 6(2): 46–49. PMID: 22511431. doi: 10.5489/cuaj.10134.