Abstracts from the Immunodeficiency Canada—7th SCID Symposium, Montreal, QC, 24 October 2019
Variable spectrum of organ involvement in siblings with LRBA deficiency
Keelia Farrella, Rae Bragerb, Thomas Issekutza, Beata Derfalvia
aDepartment of Pediatrics, Division of Immunology, IWK Health Center, Dalhousie University, Halifax, NS, Canada
bDepartment of Pediatrics, Division of Rheumatology, Immunology, and Allergy, McMaster University, Hamilton, ON, Canada
Background: LRBA (LPS responsive beige-like anchor protein) deficiency is an inborn error of immunity that was first identified in 2012 in 4 consanguineous families in which multiple members presented with early-onset hypogammaglobulinemia, infections and autoimmunity (Lopez-Herrera et al. 2012).
Cases identified since that time have demonstrated the diversity of the phenotypic manifestations of LRBA deficiency. Patients have presented with polyautoimmunity without hypogammaglobulinemia, immune dysregulation and colitis (Alangari et al. 2012; Serwas et al. 2015; Gámez-Díaz et al. 2016; Lévy et al. 2016), IPEX-like syndrome (Charbonnier et al. 2015; Gámez-Díaz et al. 2016; Eren Akarcan et al. 2018), as well as organomegaly and lymphocytic infiltration of non-lymphoid organs (Alkhairy et al. 2016; Kostel Bal et al. 2017).
There is no clear genotype-phenotype correlation and penetrance is variable: asymptomatic patients have also been reported, the oldest being 17 years old without any manifestations despite absent LRBA levels (Kostel Bal et al. 2017). A sibling with the same mutation, presented at 3 years old with autoimmune manifestations and lymphoproliferation (Kostel Bal et al. 2017).
Most patients have normal CD4+ and CD8+ T-cell immunophenotyping, with decreased numbers of Treg cells. Low B-cells are also characteristic, specifically low switched memory B-cells and low plasmablasts (Gámez-Díaz et al. 2016; Azizi et al. 2017). The term LATAIE (LRBA deficiency with autoantibodies, regulatory T (Treg) cell defects, autoimmune infiltration, and enteropathy) summarizes the most prominent phenotypic and immunological features of the disorder (Lo et al. 2016).
LRBA deficiency typically occurs due to homozygous or compound heterozygous mutations in the gene. Recently there has been a reported case of LRBA deficiency due to uniparental disomy (Soler-Palacín et al. 2018). LRBA protein transcription is induced in B-cells and macrophages by bacterial lipopolysaccharides (Wang et al. 2004). Increased LRBA expression has been seen in some cancer cell lines (Wang et al. 2004). This, and the fact that LRBA is also expressed in many non-immune cells, points to a role for LRBA in general cell growth and survival.
LRBA is known to participate in the endocytic pathway. It is important in the vesicular trafficking of CTLA-4 during its recycling from endosomes back to the cell membrane, making CTLA-4 a therapeutic target in LRBA deficiency (Lo et al. 2015). Interestingly, some patients with LRBA deficiency and autoimmunity have been shown to have normal CTLA-4 and Treg cells with increased Th17 activity (De Bruyne et al. 2018) which emphasizes the further research necessary to understand this disease. It is possible that LRBA mediates the recycling of other key receptors in endosomes that are critical to normal immune function. LRBA deficiency is also associated with impaired autophagy and increased susceptibility to apoptosis (Lopez-Herrera et al. 2012; Martínez Jaramillo and Trujillo-Vargas 2018).
Our abstract describes the cases of 3 siblings who arrived as refugees to Canada in 2017. The parents are first cousins and there was an extensive history of consanguinity among relatives. They were determined to have a new homozygous splice site mutation resulting in LRBA deficiency. The oldest sibling presented with a unique manifestation which has yet to be reported in the literature, lymphocytic infiltration of the bladder.
Clinical Features:
Sibling 1: The first sibling to present, an 11 years old female, was initially seen in December 2017 for failure to thrive, recurrent infections, chronic diarrhea and hypogammaglobulinemia.
Parents reported that she was well until the age of 3 when she began to have recurrent sinopulmonary infections, including an estimated 10 episodes of otitis media per year. She had arthritis involving her left knee since age 4 which had been treated with prednisone. Trials of methotrexate for 2 years and adalimumab for 1 year had been unsuccessful. At 4 years old she developed severe diarrhea with 6–7 watery bowel movements per day, which was ongoing at the time of her initial assessment. She also had severe failure to thrive, with a weight at 50th % for a 4.5 year old and significantly delayed bone age.
Investigations revealed low immunoglobulins with IgG <0.5 g/L, IgM 0.25 g/L, and undetectable IgA (<0.1 g/L) in the context of normal albumin. On flow cytometry she had low B-cells at 4% (just below the 5th % for her age), with 1% class-switched memory B-cells. CD4+ and CD8+ T cells were appropriately represented. NK cells were low at 2%, which is below the 5th % for her age. Specific antibody titres were not measured as the patient’s vaccination history was unclear. Lymphocyte proliferation to mitogens was normal.
Due to these findings, along with her severe symptoms, she was started on replacement immunoglobulin at 1 g/kg. Despite this, infections remained problematic. In February 2018, she developed increasing watery stools which were positive for Campylobacter. Initial treatment with oral antibiotics was unsuccessful and the patient developed Clostridium difficile. She was admitted and treated with 2 courses of IV meropenem. Unfortunately her diarrhea resumed when meropenem was discontinued. Endoscopy demonstrated chronic autoimmune/reactive inflammation in her bowel, as well as Candida esophagitis. A biopsy of the gastric antrum was positive for HHV6. The candida was successfully treated with caspofungin. In consultation with the Infectious Disease team, the HHV6 was not treated. Next Generation Sequencing of 247 primary immunodeficiency genes revealed a homozygous splice site variant mutation in LRBA at the locus c.2258+2T>G, confirming the patient’s diagnosis of LRBA deficiency.
Due to ongoing GI symptoms in the context of LRBA deficiency and previously reported success of Abatacept in this patient population, biweekly Abatacept infusion (20 mg/kg) was initiated. There was no obvious initial improvement after 6 weeks, therefore Sirolimus (1.3 mg BID) was added, and TPN was started. The diarrhea improved significantly on this regimen, as well as a maintenance dose of oral steroid. After 1 month, TPN was weaned and weight was maintained with oral supplementation. Infectious prophylaxis with daily cephalexin was added to her monthly IVIG.
Additional characteristics of LRBA deficiency in this patient included chronic CMV viremia and recurrent HSV-1 with oral ulcers. The CMV was treated with Ganciclovir but she developed ITP and neutropenia, making it unclear whether this were secondary to underlying disease or the medication. Her initial hepatomegaly improved significantly after the CMV was treated. She had significant shortness of breath, exercise intolerance, and orthopnea. A CT scan revealed multi-lobar cystic bronchiectasis. She was also noted to have hypokalemia with renal loss of both protein and potassium. This was thought to be secondary to an underlying tubulopathy, although biopsy was not performed. An echocardiogram showed a large aorta, the etiology which has not been established.
Sibling 2: The second sibling, a 13 years old male, presented with chronic recurrent abdominal pain. History was otherwise only significant for 1 episode of hematuria which resolved with antibiotics. Growth and development were normal. However during his sibling’s admission, the medical team noted that he was having recurrent abdominal pain and urinary tract infection symptoms. The pain was suprapubic, seemed positional, and significant in that he was unable to walk completely upright.
Extensive infectious investigations of urine, blood, and stool failed to isolate a pathogen. Urinalysis showed microscopic hematuria as well as 1 g/L of proteinuria. This was confirmed with a repeat urinalysis a week later. A kidney biopsy was performed which revealed thin basement membrane disease and sclerotic glomeruli. Urology was involved and initial urodynamic studies were normal. Cystoscopy showed bladder and urethral irritation. Biopsies revealed primarily T-lymphocytic infiltration, but also eosinophils, neutrophils and rare plasma cells. The biopsy was also positive for EBV but blood remained EBV negative. The ultimate pathological diagnosis was interstitial cystitis.
Immunological investigations were initiated and showed low immunoglobulins with IgG 6.29 g/L, IgM 0.08 g/L, and IgA <0.1 g/L. Albumin was normal. On flow cytometry B-cells were 1% (below the 5th %), with 3% class-switched memory B-cells. CD4+, CD8+ T cells, and NK cell numbers were normal. Titres were positive for Rubella, Measles and Hepatitis B but negative for Tetanus, Diphtheria, and Pneumococcus. However, these results were challenging to interpret given the unclear vaccination history. DNA sequencing revealed the same homozygous mutation in the LRBA gene as his sister.
Trials of Hydroxyzine and Sirolimus were both unsuccessful in reducing symptoms, which progressed to include urinary frequency. Prednisone 1 mg/kg/day and gabapentin 100 mg BID were initiated. This treatment regimen helped to lessen his symptoms significantly. He was also started on monthly IVIG.
The patient continues to have difficulty with urinary frequency, urgency, and dysuria. He has now developed pelvic floor dysynergy and has rectal pain as well. This is being managed with physiotherapy and anti-spasmodic agents. He also developed enteropathy with watery stools at age 13, and was initially treated for Campylobacter. Despite resolution of the infection, enteropathy recurred and is currently managed with Abatacept. Growth is being supported with NG feeds.
Sibling 3: The third sibling is 12 months old at the time of writing and underwent genetic testing at birth which revealed the same homozygous mutations as the elder 2 children. Thus far, failure to thrive has been the sole symptom but has been addressed with fortification of feeds.
Conclusions: This report of 3 additional cases adds to the growing phenotypic classification of LRBA deficiency. Sibling 2 presented with a manifestation of LRBA deficiency that has yet to be reported in the literature, lymphocytic infiltration of the bladder. This manifestation has had a significant deleterious impact on our patient’s functioning and quality of life. The multi-systemic involvement and complications of LRBA deficiency make treatment challenging and the involvement of a multidisciplinary team of paramount importance.
REFERENCES
Alangari, A., Alsultan, A., Adly, N., Massaad, M.J., Kiani, I.S., Aljebreen, A., Raddaoui, E., Almomen, A.K., Al-Muhsen, S., Geha, R.S., and Alkuraya, F.S. 2012. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J. Allergy Clin. Immunol. 130(2):481–488.e2. PMID: 22721650. doi: 10.1016/j.jaci.2012.05.043.
Alkhairy, O.K., Abolhassani, H., Rezaei, N., Fang, M., Andersen, K.K., Chavoshzadeh, Z., Mohammadzadeh, I., El-Rajab, M.A., Massaad, M., Chou, J., Aghamohammadi, A., Geha, R.S., and Hammarström, L. 2016. Spectrum of phenotypes associated with mutations in LRBA. J. Clin. Immunol. 36(1):33–45. PMID: 26707784. doi: 10.1007/s10875-015-0224-7.
Azizi, G., Abolhassani, H., Mahdaviani, S.A., Chavoshzadeh, Z., Eshghi, P., Yazdani, R., Kiaee, F., Shaghaghi, M., Mohammadi, J., Rezaei, N., Hammarström, L., and Aghamohammadi, A. 2017. Clinical, immunologic, molecular analyses and outcomes of Iranian patients with LRBA deficiency: A longitudinal study. Pediatr. Allergy Immunol. 28(5):478–484. PMID: 28512785. doi: 10.1111/pai.12735.
Charbonnier, L.M., Janssen, E., Chou, J., Ohsumi, T.K., Keles, S., Hsu, J.T., Massaad, M.J., Garcia-Lloret, M., Hanna-Wakim, R., Dbaibo, G., Alangari, A.A., Alsultan, A., Al-Zahrani, D., Geha, R.S., and Chatila, T.A. 2015. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J. Allergy Clin. Immunol. 135:217–227. PMID: 25468195. doi: 10.1016/j.jaci.2014.10.019.
De Bruyne, M., Bogaert, D.J., Venken, K., Van den Bossche, L., Bonroy, C., Roels, L., Tavernier, S.J., van de Vijver, E., Driessen, A., van Gijn, M., Gámez-Diaz, L., Elewaut, D., Grimbacher, B., Haerynck, F., Moes, N., and Dullaers, M. 2018. A novel LPS-responsive beige-like anchor protein (LRBA) mutation presents with normal cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and overactive TH17 immunity. J. Allergy Clin. Immunol. 142(6):1968–1971. PMID: 30193839. doi: 10.1016/j.jaci.2018.08.026.
Eren Akarcan, S., Edeer Karaca, N., Aksu, G., Aykut, A., Yilmaz Karapinar, D., Cetin, F., Aydinok, Y., Azarsiz, E., Gambineri, E., Cogulu, O., Ulusoy Severcan, E., Alper, H., and Kutukculer, N. 2018. Two male siblings with a novel LRBA mutation presenting with different findings of IPEX syndrome. JMM Case Rep. 5(10):e005167. PMID: 30479781. doi: 10.1099/jmmcr.0.005167.
Gámez-Díaz, L., August, D., Stepensky, P., Revel-Vilk, S., Seidel, M.G., Noriko, M., Morio, T., Worth, A.J.J., Blessing, J., Van de Veerdonk, F., Feuchtinger, T., Kanariou, M., Schmitt-Graeff, A., Jung, S., Seneviratne, S., Burns, S., Belohradsky, B.H., Rezaei, N., Bakhtiar, S., Speckmann, C., Jordan, M., and Grimbacher, B. 2016. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J. Allergy Clin. Immunol. 137(1):223–230. PMID: 26768763. doi: 10.1016/j.jaci.2015.09.025.
Kostel Bal, S., Haskologlu, S., Serwas, N.K., Islamoglu, C., Aytekin, C., Kendirli, T., Kuloglu, Z., Yavuz, G., Dalgic, B., Siklar, Z., Kansu, A., Ensari, A., Boztug, K., Dogu, F., and Ikinciogullari, A. 2017. Multiple presentations of LRBA deficiency: A single-center experience. J. Clin. Immunol. 37(8):790–800. PMID: 28956255. doi: 10.1007/s10875-017-0446-y.
Lévy, E., Stolzenberg, M.C., Bruneau, J., Breton, S., Neven, B., Sauvion, S., Zarhrate, M., Nitschké, P., Fischer, A., Magérus-Chatinet, A., Quartier, P., and Rieux-Laucat, F. 2016. LRBA deficiency with autoimmunity and early onset chronic erosive polyarthritis. Clin. Immunol. 168:88–93. PMID: 27057999. doi: 10.1016/j.clim.2016.03.006.
Lo, B., Zhang, K., Lu, W., Zheng, L., Zhang, Q., Kanellopoulou, C., Zhang, Y., Liu, Z., Fritz, J.M., Marsh, R., Husami, A., Kissell, D., Nortman, S., Chaturvedi, V., Haines, H., Young, L.R., Mo, J., Filipovich, A.H., Bleesing, J.J., Mustillo, P., Stephens, M., Rueda, C.M., Chougnet, C.A., Hoebe, K., McElwee, J., Hughes, J.D., Karakoc-Aydiner, E., Matthews, H.F., Price, S., Su, H.C., Rao, V.K., Lenardo, M.J., and Jordan, M.B. 2015. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 349(6246):436–440. PMID: 26206937. doi: 10.1126/science.aaa1663.
Lo, B., Fritz, J.M., Su, H.C., Uzel, G., Jordan, M.B., and Lenardo, M.J. 2016. CHAI and LATAIE: New genetic diseases of CTLA-4 checkpoint insufficiency. Blood. 128:1037–1042. PMID: 27418640. doi: 10.1182/blood-2016-04-712612.
Lopez-Herrera, G., Tampella, G., Pan-Hammarström, Q., Herholz, P., Trujillo-Vargas, C.M., Phadwal, K., Simon, A.K., Moutschen, M., Etzioni, A., Mory, A., Srugo, I., Melamed, D., Hultenby, K., Liu, C., Baronio, M., Vitali, M., Philippet, P., Dideberg, V., Aghamohammadi, A., Rezaei, N., Enright, V., Du, L., Salzer, U., Eibel, H., Pfeifer, D., Veelken, H., Stauss, H., Lougaris, V., Plebani, A., Gertz, E.M., Schäffer, A.A., Hammarström, L., and Grimbacher, B. 2012. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am. J. Hum. Genet. 90(6):986–1001. PMID: 22608502. doi: 10.1016/j.ajhg.2012.04.015.
Martínez Jaramillo, C., and Trujillo-Vargas, C.M. 2018. LRBA in the endomembrane system. Colomb. Med. 49(3):236–243. PMID: 30410199.
Serwas, N.K., Kansu, A., Santos-Valente, E., Kuloğlu, Z., Demir, A., Yaman, A., Gamez Diaz, L.Y., Artan, R., Sayar, E., Ensari, A., Grimbacher, B., and Boztug, K. 2015. Atypical manifestation of LRBA deficiency with predominant IBD-like phenotype. Inflamm. Bowel Dis. 21:40–47. PMID: 25479458. doi: 10.1097/MIB.0000000000000266.
Soler-Palacín, P., Garcia-Prat, M., Martín-Nalda, A., Franco-Jarava, C., Rivière, J.G., Plaja, A., Bezdan, D., Bosio, M., Martínez-Gallo, M., Ossowski, S., and Colobran, R. 2018. LRBA deficiency in a patient with a novel homozygous mutation due to chromosome 4 segmental uniparental isodisomy. Front. Immunol. 9:2397. PMID: 30386343. doi: 10.3389/fimmu.2018.02397.
Wang, J.W., Gamsby, J.J., Highfill, S.L., Mora, L.B., Bloom, G.C., Yeatman, T.J., Pan, T.C., Ramne, A.L., Chodosh, L.A., Cress, W.D., Chen, J., and Kerr, W.G. 2004. Deregulated expression of LRBA facilitates cancer cell growth. Oncogene. 23(23):4089–4097. PMID: 15064745. doi: 10.1038/sj.onc.1207567.
Long-term PEG-ADA enzyme replacement therapy in a patient with partial adenosine deaminase deficiency
Lucy Duana, Chaim M. Roifmana,b
aDivision of Clinical Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children and the University of Toronto, Toronto, ON, Canada
bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and the University of Toronto, Toronto, ON, Canada
Background: Adenosine deaminase (ADA) catalyzes the deamination of adenosine and deoxyadenosine to inosine and deoxyinosine, respectively, which can then be converted to waste products and excreted by the body (Grunebaum et al. 2013). ADA is a ubiquitous enzyme and activity varies in the body, the highest levels are seen in the thymus and lymphoid tissue. When ADA function is defective, there is both intracellular and extracellular accumulation of toxic substrates adenosine and deoxyadenosine, leading to abnormal cell signaling, energy production, and lymphocyte development (Grunebaum et al. 2013). ADA deficiency is an autosomal recessive primary immunodeficiency disorder of the purine salvage pathway that leads to dysfunction of T, B, and NK lymphocytes and systemic abnormalities.
ADA deficiency has an approximate incidence of 1/200 000 live births and accounts for 10%–15% of all cases of severe combined immune deficiency (SCID) (Bradford et al. 2017). Infants with ADA-deficiency present with life-threatening recurrent opportunistic infections, failure to thrive, and chronic diarrhea (Bradford et al. 2017). In addition to SCID, a group of patients present with milder, delayed onset, or late onset disease (Grunebaum et al. 2013). These patients with “partial” ADA deficiency can have variable cellular and humoral immunity that wanes. Patients with partial ADA deficiency exhibit variable degrees of immune deficiency and dysregulation (Hirschhorn and Ellenbogen 1986). Clinical features of patients described in the literature range from completely normal immune function to recurrent bacterial and viral infections and autoimmunity (Levy et al. 1988; Hirschhorn et al. 1989, 1990; Santisteban et al. 1993; Ariga et al. 2001; Artac et al. 2010).
Definitive therapy for ADA-deficiency SCID includes gene therapy and hematopoietic stem cell transplantation (HSCT) (Kohn et al. 2019). Enzyme replacement therapy (ERT) with polyethylene glycol conjugated adenosine deaminase (PEG-ADA) is used as an immediate stabilizing intervention, and in most cases used for relatively short periods (Grunebaum et al. 2013; Kohn et al. 2019). Management for patients with partial ADA deficiency is less well characterized given the rarity of the condition and wide variability of patient presentations (Santisteban et al. 1993). Antibiotic prophylaxis and immune globulin replacement should be considered in patients with abnormal immune function, and some patients are treated with PEG-ADA, although long-term treatment data is not well described in the literature (Levy et al. 1988; Santisteban et al. 1993). We report an unusual presentation of a patient with partial ADA deficiency, who was well until 5 months of age where he presented with recurrent infections. His clinical course was complicated by both autoimmune hepatitis and lung inflammation that improved with long-term PEG-ADA ERT.
Methods: Clinical and laboratory information was collected by retrospective chart review. Informed consent for the case report was obtained.
Results:
Clinical features at presentation: The patient is a 17-year-old male of Somalian descent, born to non-consanguineous parents. At 5–6 weeks of age, he developed a perianal abscess that required surgical drainage. Between 6 and 17 months of age, he had multiple episodes of sinusitis and pneumonia. At 17 months of age, he developed streptococcus pneumonia and bacteremia and herpes simplex virus (HSV)-1 induced oral ulcers, requiring acyclovir. Around 18 months of age, he presented with scleral icterus and pruritus. Laboratory investigations at the time demonstrated elevated liver enzymes consistent with hepatitis. Shortly after, the patient developed respiratory distress and a lung biopsy demonstrated inflammatory infiltrates consisting of plasma cells, eosinophils, and lymphocytes.
Immunological and genetic investigations: When the patient was referred to our institution at 2 years of age, his initial investigations demonstrated low numbers of T, B, and NK lymphocytes with elevated IgG (Table 1). Lymphocyte response to mitogen stimulation was also significantly reduced, suggesting combined immunodeficiency. However, ultrasound of the thymus revealed a normal-sized gland. His T-cell Vβ repertoire was normal. ADA level was 4 nmol/minute/mL and PNP level was 2221 nmol/minute/mL. ADA enzyme activity in the patient’s erythrocytes and lymphocytes was 3% compared with healthy controls. His parents were found to have one third and half of the enzymatic activity compared with controls.
Table 1:
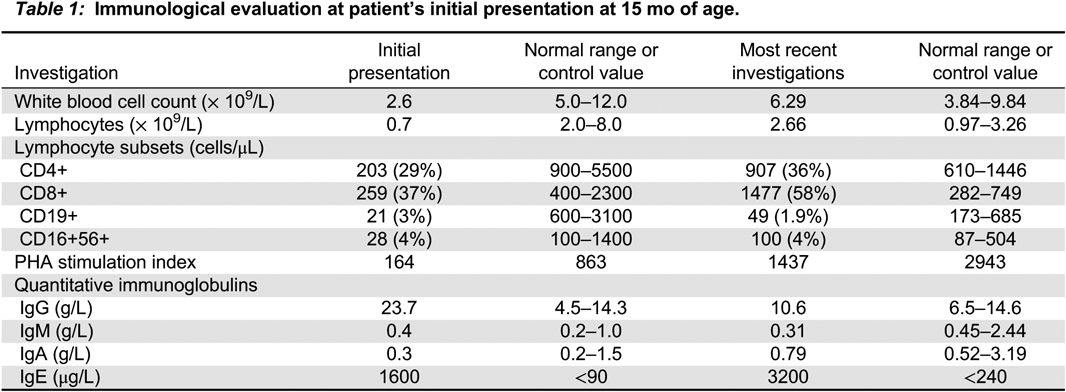
Direct sequencing of the ADA gene identified 2 missense mutations (Santisteban et al. 1993, 1995; Somech et al. 2009).
He was diagnosed with partial ADA deficiency, and started on PEG-ADA ERT at 23 months of age and Pneumocystis jiroveci prophylaxis with tovaquone. Gradual improvement of respiratory symptoms was observed over 4–5 months. Following diagnosis of hepatitis, further investigations showed elevated anti-liver microsomal antibody titre 1:640 and positive anti-smooth muscle antibody titre 1:32. A liver biopsy demonstrated portal tract infiltration with eosinophils and plasma cells. Both findings were suggestive of an underlying autoimmune process. Prednisone was initiated at a dose of 1 g/kg/day. The combination of ERT and steroids lead to decrease in the patient’s liver enzymes and steroids were weaned off over 3 months. In regards to consideration for definitive therapy, his diagnosis of partial ADA deficiency was not an indication for HSCT, he did not have an appropriate donor even if HSCT was indicated, and gene therapy was not available at the time.
Long-term management: PEG-ADA was discontinued at 3 years of age. About 6 months later, his lymphocyte markers and proliferation to mitogen simulation was significantly reduced, therefore, he was restarted on PEG-ADA. Repeat immunological investigations after 3 months demonstrated improved white blood cell count 5.3 × 109/L, lymphocyte count 1.82 × 109/L, and lymphocyte subsets. Lymphocyte proliferation to mitogen stimulation also showed improvement. His most recent immunological investigations demonstrate stable T-cell lymphocyte count with B-cell lymphopenia, and normal lymphocyte proliferation to mitogen and antigen stimulation (Table 1). In last 2–3 years, there had been a slow increase in toxic metabolites prior to adjustment of his PEG-ADA dose. Overall, he has remained clinically stable without significant infections or autoimmunity, despite his initial severe presentation.
Conclusions: We report a patient with partial ADA deficiency who presented in early childhood with recurrent infections, autoimmune hepatitis, and lung inflammation. Two missense mutations in the ADA gene were identified, that lead to a unique compound heterozygosity of pathogenic variants which presented as partial ADA deficiency. He has required long-term treatment with PEG-ADA ERT.
In contrast to other patients described in the literature with partial ADA deficiency, he had a more severe course in infancy that significantly improved following long-term treatment with PEG-ADA. Although autoimmune cytopenias, diabetes mellitus, and hypothyroidism has been reported in other patients with partial ADA deficiency, autoimmune hepatitis has only been characterized in our patient (Somech et al. 2009). Hepatitis has been documented in a patient with ADA-deficiency SCID (Bollinger et al. 1996).
PEG-ADA is commonly used for short periods of time before definitive HSCT or gene therapy in ADA-deficiency. There have been no systematic studies of long-term treatment with PEG-ADA and 2 studies have demonstrated partial immune restoration with long-term PEG-ADA treatment (Chan et al. 2005; Malacarne et al. 2005). Long-term treatment with PEG-ADA has been associated with infections, autoimmunity, and malignancy (Grunebaum et al. 2013; Kohn et al. 2019). There has been continued good health after 25 years of PEG-ADA reported in 1 ADA-deficiency SCID patient (Tartibi et al. 2016). Our patient currently continues on treatment with PEG-ADA and has been overall clinically well without significant infections. It is important to closely monitor his clinical status and immunological work-up, and consider if and when definitive therapy may be required as well as the risks and benefits of the options.
REFERENCES
Ariga, T., Oda, N., Sanstisteban, I., Arredondo-Vega, F.X., Shioda, M., Ueno, H., Terada, K., Kobayashi, K., Hershfieldm, M.S., and Sakiyama, Y. 2001. Molecular basis for paradoxical carriers of adenosine deaminase (ADA) deficiency that show extremely low levels of ADA activity in peripheral blood cells without immunodeficiency. J. Immunol. 166(3):1698–1702. PMID: 11160213. doi: 10.4049/jimmunol.166.3.1698.
Artac, H., Göktürk, B., Bozdemir, S.E., Toy, H., Van Der Burg, M., Santisteban, I., Hershfield, M., and Reisli, I. 2010. Late-onset adenosine deaminase deficiency presenting with Heck’s disease. Eur. J. Pediatr. 169(8):1033–1036. PMID: 20039061. doi: 10.1007/s00431-009-1131-9.
Bollinger, M.E., Arredondo-Vega, F.X., Santisteban, I., Schwarz, K., Hershfield, M.S., and Lederman, H.M. 1996. Brief report: Hepatic dysfunction as a complication of adenosine deaminase deficiency. N. Engl. J. Med. 334(21):1367–1371. PMID: 8614422. doi: 10.1056/NEJM199605233342104.
Bradford, K.L., Moretti, F.A., Carbonaro-Sarracino, D.A., Gaspar, H.B., and Kohn, D.B. 2017. Adenosine deaminase (ADA)-deficient severe combined immune deficiency (SCID): Molecular pathogenesis and clinical manifestations. J. Clin. Immunol. 37(7):626–637. PMID: 28842866. doi: 10.1007/s10875-017-0433-3.
Chan, B., Wara, D., Bastian, J., Hershfield, M.S., Bohnsack, J., Azen, C.G., Parkman, R., Weinberg, K., and Kohn, D.B. 2005. Long-term efficacy of enzyme replacement therapy for adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID). Clin. Immunol. 117(2):133–143. PMID: 16112907. doi: 10.1016/j.clim.2005.07.006.
Grunebaum, E., Cohen, A., and Roifman, C.M. 2013. Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies. Curr. Opin. Allergy Clin. Immunol. 13(6):630–638. PMID: 24113229. doi: 10.1097/ACI.0000000000000006.
Hirschhorn, R., and Ellenbogen, A. 1986. Genetic heterogeneity in adenosine deaminase (ADA) deficiency: Five different mutations in five new patients with partial ADA deficiency. Am. J. Hum. Genet. 38(1):13–25. PMID: 3946419.
Hirschhorn, R., Tzall, S., Ellenbogen, A., and Orkin, S.H. 1989. Identification of a point mutation resulting in a heat-labile adenosine deaminase (ADA) in two unrelated children with partial ADA deficiency. J. Clin. Invest. 83(2):497–501. PMID: 2783588. doi: 10.1172/JCI113909.
Hirschhorn, R., Tzall, S., and Ellenbogen, A. 1990. Hot spot mutations in adenosine deaminase deficiency. Proc. Natl. Acad. Sci. 87(16):6171–6175. PMID: 2166947. doi: 10.1073/pnas.87.16.6171.
Kohn, D.B., Hershfield, M.S., Puck, J.M., Aiuti, A., Blincoe, A., Gaspar, H.B., Notarangelo, L.D., and Grunebaum, E. 2019. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J. Allergy Clin. Immunol. 143(3):852–863. PMID: 30194989. doi: 10.1016/j.jaci.2018.08.024.
Levy, Y., Hershfield, M.S., Fernandez-Mejia, C., Polmar, S.H., Scudiery, D., Berger, M., and Sorensen, R.U. 1988. Adenosine deaminase deficiency with late onset of recurrent infections: Response to treatment with polyethylene glycol-modified adenosine deaminase. J. Pediatr. 113(2):312–317. PMID: 3260944. doi: 10.1016/s0022-3476(88)80271-3.
Malacarne, F., Benicchi, T., Notarangelo, L.D., Mori, L., Parolini, S., Caimi, L., Hershfield, M., Notarangelo, L.D., and Imberti, L. 2005. Reduced thymic output, increased spontaneous apoptosis and oligoclonal B cells in polyethylene glycol-adenosine deaminase-treated patients. Eur. J. Immunol. 35(11):3376–3386. PMID: 16276484. doi: 10.1002/eji.200526248.
Santisteban, I., Arredondo-Vega, F.X., Kelly, S., Mary, A., Fischer, A., Hummell, D.S., Lawton, A., Sorensen, R.U., Stiehm, E.R., and Uribe, L. 1993. Novel splicing, missense, and deletion mutations in seven adenosine deaminase-deficient patients with late/delayed onset of combined immunodeficiency disease. Contribution of genotype to phenotype. J. Clin. Invest. 92(5):2291–2302. PMID: 8227344. doi: 10.1172/JCI116833.
Santisteban, I., Arredondo-Vega, F.X., Kelly, S., Loubser, M., Meydan, N., Roifman, C., Howell, P.L., Bowen, T., Weinberg, K.I., Schroeder, M.L., and Hershfield, M.S. 1995. Three new adenosine deaminase mutations that define a splicing enhancer and cause severe and partial phenotypes: Implications for evolution of a CpG hotspot and expression of a transduced ADA cDNA. Hum. Mol. Genet. 4(11):2081–2087. PMID: 8589684. doi: 10.1093/hmg/4.11.2081.
Somech, R., Lai, Y.H., Grunebaum, E., Le Saux, N., Cutz, E., and Roifman, C.M. 2009. Polyethylene glycol-modified adenosine deaminase improved lung disease but not liver disease in partial adenosine deaminase deficiency. J. Allergy Clin. Immunol. 124(4):848–850. PMID: 19665771. doi: 10.1016/j.jaci.2009.07.003.
Tartibi, H.M., Hershfield, M.S., and Bahna, S.L. 2016. A 24-year enzyme replacement therapy in an adenosine-deaminase-deficient patient. Pediatrics. 137(1):e20152169. PMID: 26684479. doi: 10.1542/peds.2015-2169.
Survival of patients with severe combined immunodeficiency in the newborn screening era: a single-centre experience 2013–2019
Ori Scotta, Rae Bragerb, Peter Dentb, Victoria Siuc, Brenda Reida, Vy Hong-Diep Kima, Chaim M. Roifmana
aDivision of Clinical Immunology and Allergy, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
bDivision of Clinical Immunology and Allergy, McMaster University, Hamilton, ON, Canada
cDivision of Medical Genetics, London Health Sciences Centre, London, ON, Canada
Background: Severe combined immunodeficiency (SCID) is marked by profoundly diminished T-cell immunity and predisposition to viral, bacterial, fungal and opportunistic infections. The gold standard for treating SCID is allogenic hematopoietic stem cell transplant (HSCT), without which SCID is invariably fatal within the first 2 years of life (Dalal et al. 2000). In this regard, a long-standing notion has been the importance of early intervention, which was believed to lead to better outcome. A seminal study from Duke University has established 3.5 months as an important age cut-off affecting transplant outcome. Infants transplanted prior to this age were typically free of infection at the time of transplant, and fared better at 5 years compared to those transplanted at a later age, with >90% survival (Buckley 2004). Moreover, fiscal reports have determined that the cost of care for SCID patient is significantly higher following late transplant (after 3.5 months) compared with early transplant (Clément et al. 2015).
In an effort to promote early identification and treatment of SCID patients, the T-cell receptor excision circle (TREC) test has been gradually introduced into newborn screening (NBS) programs across North American and Europe. TRECs are short segments of DNA excised from the T-cell receptor upon maturation in the thymus, and serve as a surrogate marker for T-cell development. The NBS TREC assay utilizes extracted DNA from dried blood spot, which undergoes quantitative real time PCR to determine the extent of TREC per microliter (Douek et al. 2000; Chan and Puck 2005; Puck 2011). The province of Ontario added TREC assay to its NBS panel in 2013, along with the establishment of a system for result flagging, notification and follow-up (Figure 1; Reid et al. 2017). This report reviews the survival of 14 children with SCID, diagnosed and treated at our centre between 2013 and 2019.
Figure 1:
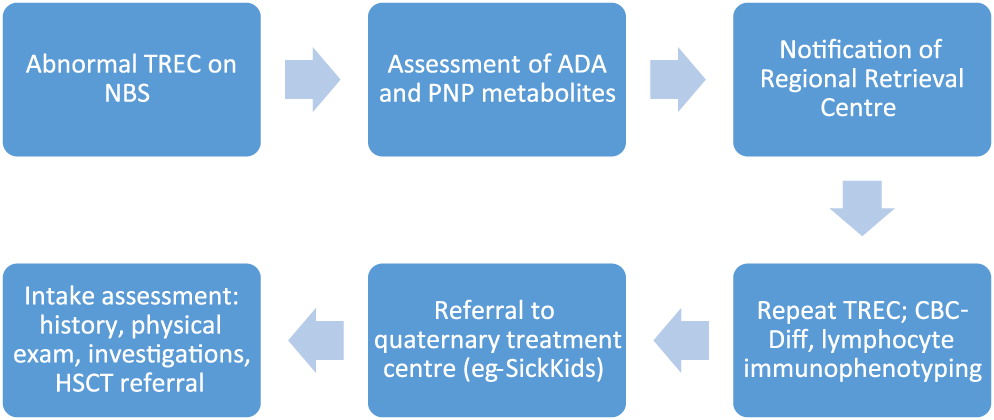
Methods: We performed a chart review of patients diagnosed with SCID, and treated by our centre since the introduction of TREC into NBS. The diagnosis of SCID followed the PIDTC criteria, defined by presence of <300 autologous CD3+ cells/µL, or <200 naïve CD4+ cells/µL. The definition of leaky SCID with Omenn syndrome mandated at least 300 autologous CD3+ cells/µL, and the presence of autoreactive oligoclonal T-cells, rash, and eosinophilia.
Results: Between 2013 and 2019, 14 infants were diagnosed with SCID and received treatment at the Hospital for Sick Children. All patients had an abnormal TREC assay on NBS, and 13 were identified prior to manifesting symptoms. Unfortunately, 1 patient was not flagged for notification by the NBS centre, and presented with disseminated infection at the age of 7 months. All other patients were assessed by our service within the first 3 weeks of life. No false negative TREC assays have been reported in any of our SCID patients. The most commonly identified cause of SCID was adenosine deaminase (ADA) deficiency, followed by interleukin 2 receptor gamma (IL2Rγ) deficiency. A genetic diagnosis was not determined in 4 patients, of whom 2 presented with leaky SCID. For a breakdown of patient diagnosed, please refer to Figure 2.
Figure 2:
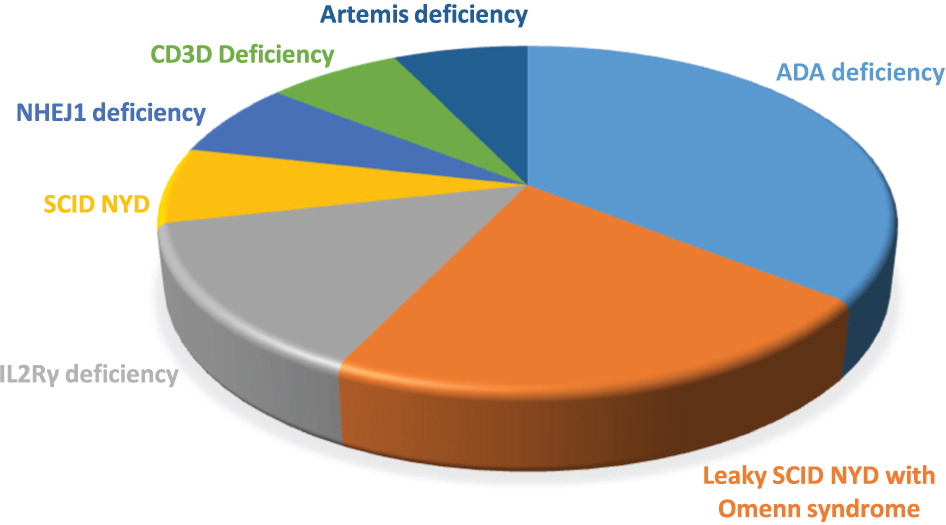
Stupendous survival has been noted among ADA-deficient patients in our cohort, all of whom underwent gene therapy (GT) between 7 and 15 months of age. All are alive, and most have shown encouraging immune reconstitution. Among 9 patients with non-ADA deficient SCID, 8 were transplanted between 2 and 7 months of life. The patient who presented at 7 months passed away of infection prior to transplant. Prior to HSCT, 3 patients developed Omenn syndrome, which required treatment with immunosuppression. One of these patients also developed a bacterial infection requiring antimicrobials. Two patients developed viral upper respiratory tract infections prior to transplant (Parainfluenza and Entero-rhinovirus). While the infections resolved symptomatically, 1 patient suffered reactivation of her Parainfluenza infection, eventually contributing to her demise following transplant. Most patients received umbilical cord stem cells, compared to 2 who had a matched related donor. Following transplant, 3 patients have died, all of infectious causes and within the first 8 months. At present, 2 patients show good immune reconstitution and are off immunosuppression. One boy remains on long-term immunosuppression for chronic liver graft-versus-host-disease (GvHD).
To determine the potential impact of NBS in our hospital, we compared the current survival rates to those published previously by our centre in 2006 (Grunebaum et al. 2006). Between 1990 and 2004, SCID patients transplanted at the Hospital for Sick Children [HSCT from matched related donors (MRD) and matched unrelated donors (MUD)], demonstrated an overall survival rate of 83%. This appears to be no different than the 77% overall survival of SCID patients in the current cohort. However, when further comparing the 2 cohorts according to diagnosis and treatment modality, the differences between ADA-deficiency and other forms of SCID is striking. Among ADA-deficient patients, remarkable survival enhancement is seen following gene therapy compared to our previously transplanted patients (from 67% to 100% survival). In contrast, survival rates for non-ADA deficient SCID have not improved following transplant; while 85% survival was reported by our centre in 2006, survival rate in the current cohort post-HSCT has been 62.5% (Figure 3).
Figure 3:
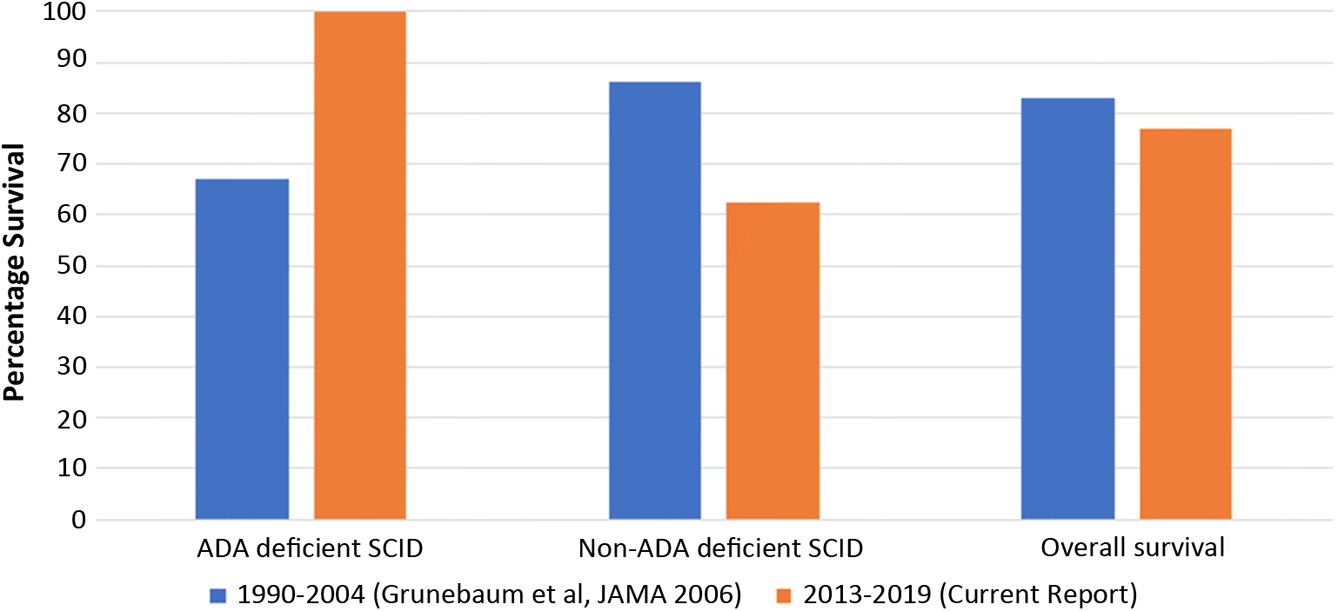
Discussion: The aim of adding TREC assessment to NBS was to enhance early detection and outcome in SCID patients. Our current cohort collected over 5 years, has shown overall improved survival among ADA-deficient patients. This is likely owing to the introduction of gene therapy, which has been performed well beyond 3.5 months of age in all of our patients. However, a similar trend of increased survival has not been noted among non-ADA deficient SCID patients.
Our results should be analyzed in the context of the experience of other centres. A recent study from California has reported that of 50 identified patients over 7 years, 49 were transplanted and 46 (94%) are alive at 1–8 years of age (Amatuni et al. 2019). Interestingly, not all patients in the California study were transplanted before 3.5 months. However, the California protocol mandates immediate admission to hospital and protective isolation upon diagnosis of SCID. Consequently, they report on very low occurrence of infection prior to transplant. The importance of infection status, rather than age at transplant, has also recently been demonstrated in a large post-transplant SCID cohort compiled by the Primary Immune Deficiency Treatment Consortium (PIDTC). Patients transplanted at <3.5 months of age (regardless of infection status) had a 2-year overall survival of 92%, compared with 96% for patients transplanted at >3.5 months of age who were infection-free at time of transplant (Heimall et al. 2017). In our centre practices have changed in recent years, and it is possible that this may factor in to the rates of infection and survival seen in our patients.
In terms of time to transplant, only 3 of our patients were transplanted prior to 3.5 months. In 5 children, a combination of medical, familial and psychosocial circumstances led to HSCT beyond 3.5 months. However, it is unclear whether the timing of transplant in our patients had a significant impact on outcome, as mortality occurred among the group of infants transplanted before 3.5 months as well.
A significant limitation of this report is the small number of patients included, which precludes us from deriving any statistically significant conclusions. However, observation of trends in the interim may be invaluable for quality assurance of this relatively new process. Certainly, data must continue to be collected and analyzed over time. Moreover, collaborating with other centres and provinces in assessing the national Canadian experience will be essential in fine-tuning the NBS process and determining other factors which impact patient outcome.
REFERENCES
Amatuni, G.S., Currier, R.J., Church, J.A., Bishop, T., Grimbacher, E., Nguyen, A.A., Agarwal-Hashmi, R., Aznar, C.P., Butte, M.J., Cowan, M.J., Dorsey, M.J., Dvorak, C.C., Kapoor, N., Kohn, D.B., Markert, M.L., Moore, T.B., Naides, S.J., Sciortino, S., Feuchtbaum, L., Koupaei, R.A., and Puck, J.M. 2019. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California, 2010–2017. Pediatrics. 143(2):e20182300. PMID: 30683812. doi: 10.1542/peds.2018-2300.
Buckley, R.H. 2004. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu. Rev. Immunol. 22:625–655. PMID: 15032591. doi: 10.1146/annurev.immunol.22.012703.104614.
Chan, K., and Puck, J.M. 2005. Development of population-based newborn screening for severe combined immunodeficiency. J. Allergy Clin. Immunol. 115(2):391–398. PMID: 15696101. doi: 10.1016/j.jaci.2004.10.012.
Clément, M.C., Mahlaoui, N., Mignot, C., Le Bihan, C., Rabetrano, H., Hoang, L., Neven, B., Moshous, D., Cavazzana, M., Blanche, S., Fischer, A., Audrain, M., and Durand-Zaleski, I. 2015. Systematic neonatal screening for severe combined immunodeficiency and severe T-cell lymphopenia: Analysis of cost-effectiveness based on French real field data. J. Allergy Clin. Immunol. 135(6):1589–1593. PMID: 25840725. doi: 10.1016/j.jaci.2015.02.004.
Dalal, I., Reid, B., Doyle, J., Freedman, M., Calderwood, S., Saunders, F., and Roifman, C.M. 2000. Matched unrelated bone marrow transplantation for combined immunodeficiency. Bone Marrow Transplant. 25(6):613–621. PMID: 10734295. doi: 10.1038/sj.bmt.1702215.
Douek, D.C., Vescio, R.A., Betts, M.R., Brenchley, J.M., Hill, B.J., Zhang, L., Berenson, J.R., Collins, R.H., and Koup, R.A. 2000. Assessment of thymic output in adults after haematopoietic stem-cell transplantation and prediction of T-cell reconstitution. Lancet. 355(9218):1875–1881. PMID: 10866444. doi: 10.1016/S0140-6736(00)02293-5.
Grunebaum, E., Mazzolari, E., Porta, F., Dallera, D., Atkinson, A., Reid, B., Notarangelo, L.D., and Roifman, C.M. 2006. Bone marrow transplantation for severe combined immune deficiency. JAMA. 295(5):508–518. PMID: 16449616. doi: 10.1001/jama.295.5.508.
Heimall, J., Logan, B.R., Cowan, M.J., Notarangelo, L.D., Griffith, L.M., Puck, J.M., Kohn, D.B., Pulsipher, M.A., Parikh, S., Martinez, C., Kapoor, N., O’Reilly, R., Boyer, M., Pai, S.Y., Goldman, F., Burroughs, L., Chandra, S., Kletzel, M., Thakar, M., Connelly, J., Cuvelier, G., Davila Saldana, B.J., Shereck, E., Knutsen, A., Sullivan, K.E., DeSantes, K., Gillio, A., Haddad, E., Petrovic, A., Quigg, T., Smith, A.R., Stenger, E., Yin, Z., Shearer, W.T., Fleisher, T., Buckley, R.H., and Dvorak, C.C. 2017. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: A PIDTC natural history study. Blood. 130(25):2718–2727. PMID: 29021228. doi: 10.1182/blood-2017-05-781849.
Puck, J.M. 2011. The case for newborn screening for severe combined immunodeficiency and related disorders. Ann. N. Y. Acad. Sci. 1246:108–117. PMID: 22236435. doi: 10.1111/j.1749-6632.2011.06346.x.
Reid, B., Ovadia, A., and Schejter, Y.D. 2017. Managing newborn screening for SCID in a referral centre. LymphoSign J. 4(2):77–79. doi: 10.14785/lymphosign-2017-0005.
Intermittent hypereosinophilia in a pediatric patient with X-linked hyper-IgM syndrome
Shama Suda, Thomas Eiweggerb, Julia Uptonc, Chaim M. Roifmand
aDivision of Immunology and Allergy, Department of Pediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
bDivision of Immunology and Allergy, Food Allergy and Anaphylaxis Program, The Hospital for Sick Children, Departments of Pediatrics and Immunology, University of Toronto, Toronto, ON, Canada
cDivision of Immunology and Allergy, Food Allergy and Anaphylaxis Program, Translational Medicine Program, Research Institute, The Hospital for Sick Children, Departments of Pediatrics and Immunology, University of Toronto, Toronto, ON, Canada
dDivision of Immunology and Allergy, Department of Pediatrics, Canadian Centre for Primary Immunodeficiency and the Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
Background: X-linked hyper-IgM syndrome (XHIGM) is a rare primary immunodeficiency in males, caused by a mutation in the CD40 ligand gene located at Xq26.3-27. The product of the CD40 ligand gene is a 39-kDa type II transmembrane glycoprotein transiently expressed on the surface of activated CD4+ T cells (Yazdani et al. 2019).
CD40 is expressed on B cells, dendritic cells, macrophages and monocytes and interacts with CD40 ligand to stimulate B cell proliferation, growth, somatic hypermutation and isotype switching. This interaction and its downstream effects are key in the production of memory B cells and working antibodies. The failure of immunoglobulin isotype switching in B cells leads to normal or elevated levels of IgM with low or absent levels of IgG, IgA and IgE. Defective CD40-CD40 ligand signaling also causes impaired IL-12 secretion from dendritic cells leading to decreased IFN-γ production from T cells. This causes impaired macrophage activation and increases the risk of opportunistic infections (Qamar and Fuleihan 2014).
Patients with XHIGM are at increased risk of infections (sino-pulmonary and opportunistic), autoimmune disease, neutropenia and malignancy. Treatment with immunoglobulin replacement is required to reduce the severity and frequency of infections. Bacterial prophylaxis is vital to prevent infection with Pneumocystis jiroveci. Patients should be treated aggressively with antimicrobial therapy when infections arise. Immune-suppressive therapy can be considered for patients with autoimmune manifestations. Bone marrow transplantation is another treatment option, particularly for patients with a fully matched donor (Yazdani et al. 2019).
Pneumocystis jiroveci is a relatively common infection in patients with CD40 ligand deficiency, often occurring in the first year of life. A report from the NIH North American USIDNET registry found that 42/132 (aged 2 months—62 years) of patients had been affected at some point in their lifetime (Leven et al. 2016).
We report the case of a 4-year-old male diagnosed with XHIGM, on trimethoprim-sulfamethoxazole prophylaxis, who presented with intermittent hypereosinophilia and was ultimately diagnosed with Pneumocystis jiroveci pneumonia and dientamoeba fragilis.
Methods: Clinical data was gathered by retrospective chart review. Informed consent was obtained from the family.
Results:
Clinical features: The patient is a 4-year-old male born to non-consanguineous parents of Caucasian background. He was diagnosed shortly after birth on the basis of genetic testing (GeneDx) on cord blood. His mother is a known carrier and had been tested after his older half-brother was diagnosed with XHIGM at 7 years of age. She was seen in the immunology clinic at the Hospital for Sick Children antenatally. There were no complications during the pregnancy and the patient was born at 38 weeks via a planned C-section. He also had a healthy half-sister. There was a maternal cousin with XHIGM who died of brain cancer at the age of 30. He was started on IVIG at 2 months of life as well as trimethoprim-sulfamethoxazole for PJP prophylaxis 3 times per week. The family was advised to avoid live viral vaccines.
He never had any significant infections or hospital admissions. He had been diagnosed with autism and also had a history of constipation.
Immunological features: Investigations done at 3 months of age, prior to starting IVIG, showed normal immunoglobulin levels (IgG 3.5, IgA <0.1 and IgM 0.1). Lymphocyte immunophenotyping showed normal T, B and NK cell numbers (CD3 6260, CD4 4612, CD8 1555, CD19 1052, NK 356, CD4:CD8 3.0). His lymphocyte proliferation test was appropriate at 609 (normal >200). Genetic testing revealed a mutation in keeping with XHIGM (Homozygous c.761 C>T p.Thx254Met (T254M)).
Hypereosinophilia: The patient had ongoing low-grade eosinophilia 0.5–1 × 109/L (normal 0.03–0.53 × 109/L) at baseline. He developed significant hypereosinophilia in June 2018 at 7.18 × 109/L noted incidentally on routine pre-IVIG bloodwork. He was urgently assessed and noted to be clinically asymptomatic with no fevers. Evaluation for end organ dysfunction of the liver (ALT 19 (normal <44), AST 33 (normal <52)) and heart (troponin 10.2 (normal <30.9 ng/L)) were unremarkable A chest X-ray was normal and he had a negative work-up for parasitic infections with 3 stool ova + parasite collections. Serology could not be performed given that the patient is on IVIG and does not make appropriate antibodies.
In August 2018, he had a bone marrow biopsy done to rule out an underlying malignancy which was negative. At that time his eosinophils had normalized without any intervention and he clinically remained asymptomatic. In December 2018 his eosinophils suddenly increased to 9.67 × 109/L. Repeat bloodwork in January 2019 showed a further increase to 25.58 × 109/L. He was admitted to hospital for further investigations. A chest X-ray showed non-specific bilateral interstitial opacification. A CT chest showed diffuse abnormal opacification in both lungs with moderate lymphadenopathy of the mediastinum suggestive or infection or inflammation. A bronchoalveolar lavage (BAL) identified Pneumocystis jiroveci pneumonia (PJP) as well as Streptococcus pneumoniae. Prior to initiation of treatment his eosinophils had dropped to 13.45 × 109/L. Treatment with IV trimethoprim-sulfamethoxazole and ceftriaxone were initiated as an inpatient then stepped down to oral treatment as an outpatient. He did not require treatment with steroids. He remained asymptomatic throughout his admission. His eosinophils have remained within the normal range since.
As per parental report although tremendous efforts were made to encourage him to take his trimethoprim-sulfamethoxazole prophylaxis prior to him developing hypereosinophilia they often had difficulty getting him to take the complete dose.
While admitted to hospital in January a repeat stool examination for O + P came back positive for dientamoeba fragilis with pus. He was prescribed metronidazole after discharge. It is unclear what his eosinophils were at initiation of treatment as they were not checked for 1 week prior to him going home (at which point they were 4.30 × 109/L).
Conclusions: We report a pediatric patient with XHIGM with intermittent hypereosinophilia who was found to have PJP on BAL as well as stool-borne parasite. It is interesting that our patient’s eosinophil count normalized spontaneously for a period of time. Whether this could be explained by possible intermittent compliance with his trimethoprim-sulfamethoxazole prophylaxis or the coinfection with dientamoeba fragilis remains unresolved.
The increased risk of Pneumocystis in patients with XHIGM has been well described. The absence of CD40L binding to CD40 expressed on dendritic cells and macrophages prevents their activation, maturation and the secretion of cytokines, all of which are necessary for an effective CD4+ T cell response (Qamar and Fuleihan 2014).
There are few case reports of a patient with XHIGM presenting with hypereosinophilia presumed to be respiratory in etiology. One case report described a patient with XHIGM with ARDS and an eosinophil count of 18%–25% that responded to treatment with trimethoprim-sulfamethoxazole (Guo et al. 2015). Another described a patient with XHIGM admitted to the ICU with pneumonia, ARDS and found to have an elevated eosinophil count. It is unclear whether this patient was treated with antibiotics (Merchant et al. 2014). Neither of these patients had a BAL performed and PJP was not isolated in either case. A third patient who presented with respiratory distress and hypereosinophilia had a negative BAL on 2 occasions for PJP but ultimately had a positive lung biopsy on silver stain (Joseph et al. 2005).
Hypereosinophilia and transaminitis has also been described as a presenting feature of sclerosing cholangitis and Cryptosporidium in patients with autosomal-recessive CD40 deficiency (Lougaris et al. 2005). To our knowledge there have been no reports of this occurring as a result of dientamoeba fragilis in XHIGM despite it being a parasite that is known to cause hypereosinophilia (Miguel et al. 2018). Furthermore, it would be unusual for any infectious etiology to cause intermittent hypereosinophilia. In patients with XHIGM presenting with hyperoesinophilia a broad differential diagnosis should be considered.
REFERENCES
Guo, L., Chen, B., Xu, B., Lu, M., Ning, B., and Chen, Z. 2015. X-linked hyper-IgM syndrome with eosinophilia in a male child: A case report. Exp. Ther. Med. 9(4):1328–1330. PMID: 25780430. doi: 10.3892/etm.2015.2261.
Joseph, L., Rudensky, B., Cohen, S., Goldberg, S., Schlesinger, Y., and Picard, E. 2005. Eosinophilia, pneumonia and hypogammaglobulinemia. Pediatr. Infect. Dis. J. 24(9):848. PMID: 16148861. doi: 10.1097/01.inf.0000178069.55725.a1.
Leven, E.A., Maffucci, P., Ochs, H.D., Scholl, P.R., Buckley, R.H., Fuleihan, R.L., Geha, R.S., Cunningham, C.K., Bonilla, F.A., Conley, M.E., Ferdman, R.M., Hernandez-Trujillo, V., Puck, J.M., Sullivan, K., Secord, E.A., Ramesh, M., and Cunningham-Rundles, C. 2016. Hyper IgM syndrome: A report from the USIDNET Registry. J. Clin. Immunol. 36(5):490–501. PMID: 27189378. doi: 10.1007/s10875-016-0291-4.
Lougaris, V., Badolato, R., Ferrari, S., and Plebani, A. 2005. Hyper immunoglobulin M syndrome due to CD40 deficiency: Clinical, molecular, and immunological features. Immunol. Rev. 203(1):48–66. PMID: 15661021. doi: 10.1111/j.0105-2896.2005.00229.x.
Merchant, R.H., Ahmed, J., Ahmed, N., and Picard, C. 2014. Type I hyper IgM syndrome with novel mutation from India. Indian J. Pediatr. 81(6):620–622. PMID: 23604614. doi: 10.1007/s12098-013-1029-4.
Miguel, L., Salvador, F., Sulleiro, E., Sánchez-Montalvá, A., Molina-Morant, D., López, I., and Molina, I. 2018. Clinical and epidemiological characteristics of patients with Dientamoeba fragilis infection. Am. J. Trop. Med. Hyg. 99(5):1170–1173. PMID: 30328410. doi: 10.4269/ajtmh.18-0433.
Qamar, N., and Fuleihan, R.L. 2014. The hyper IgM syndromes. Clin. Rev. Allergy Immunol. 46(2):120–130. PMID: 23797640. doi: 10.1007/s12016-013-8378-7.
Yazdani, R., Fekrvand, S., Shahkarami, S., Azizi, G., Moazzami, B., Abolhassani, H., and Aghamohammadi, A. 2019. The hyper IgM syndromes: Epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin. Immunol. 198:19–30. PMID: 30439505. doi: 10.1016/j.clim.2018.11.007.
Novel genetic cause of autoimmune lymphoproliferative syndrome
Amarilla B. Mandolaa,b, Nigel Sharfea,b, Chaim M. Roifmana,b
aThe Division of Immunology and Allergy, Department of Pediatrics, Hospital for Sick Children and the University of Toronto, Toronto, ON, Canada
bThe Canadian Centre for Primary Immunodeficiency and the Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, Hospital for Sick Children, Toronto, ON, Canada
Background: Autoimmune lymphoproliferative syndrome (ALPS) is characterized by defective lymphocyte apoptosis and programmed cell death which leads to immune dysregulation. The well described clinical manifestations include lymphadenopathy, hepatosplenomegaly, and multi-lineage cytopenias as well as an increased risk of lymphoma. Most cases have been found to have genetic aberrations of the FAS pathway. However, in some clinically identical patients a similar genetic abnormality cannot be identified. Diagnosis of ALPS in those patients is based on typical clinical manifestations as well as several laboratory markers such as elevated double negative T cell numbers (DNT), elevated IL10 and vitB12.
We describe here 2 patients with ALPS who were products of unrelated families. In both, we discovered novel NFKB2 mutations.
Patient Description:
Patient 1: Patient 1 is a 10 year old male born to a non-consanguineous family of Irish origin. He presented at age 3 years with immune mediated cytopenias, diffuse lymphadenopathy, hepatosplenomegaly, duodenitis, eczema and short stature. Immunological evaluation showed progressive CD4+ T cell and NK cell lymphopenia, with borderline PHA and decreased proliferation responses to CD3 mitogen stimulation. TCRV beta repertoire showed elevation in CD4+ V beta 13.1, 13.6 and CD8+ V beta 8 and 9 clones, as well underrepresentation of CD8+ V beta 5.3, 11 and 12 families. He has hypogammaglobulinemia and unsustained specific antibody titers. His double negative T cell number is between 1.8% and 4.56%.
Patient 2: Patient 2 is a currently a 32 year old male, born to non-consanguineous parents of Italian origin. He initially presented at age 2 year with Evan’s syndrome, then developed diffuse lymphadenopathy, hepatosplenomegaly, lymphocytic interstitial pneumonitis, suffered from recurrent viral infections and otitis and was worked up for congenital opsoclonus myoclonus. Immunological evaluation showed progressive and profound CD4+ and CD8+ lymphopenia, decreased NK cell numbers, hypogammaglobulinemia, and low unresponsive vaccine titers. His lymphocyte proliferation responses to PHA and mitogen stimulation progressively decreased. TCRV beta repertoire shows elevated expression of V beta CD4+ Vb2, Cd8+ Vb3, 7.1 and 13.2 clones and underrepresentation of CD4+ V beta 23 and CD8+ V beta 7.2, 12, 20 and 23 families. His double negative T cell number is between 2.5% and 5.4%.
Further assessment has demonstrated a defect in the NFKB pathway, consistent with abnormal NFKB2 signaling.
Discussion: Autoimmune lymphoproliferative syndrome is a complex disease that includes patients, that their genetic diagnosis was not established with targeted gene sequencing only by whole exome sequencing. Hereby we describe 2 patients with different ethnicity, with a novel heterozygous mutation in NFKB2, presenting with ALPS clinical phenotype (diffuse lymphadenopathy, hepatosplenomegaly, increased double negative T cells). We have demonstrated here the NFKB pathway defects may present as ALPS syndrome.
REFERENCES
Brue, T., Quentien, M.H., Khetchoumian, K., Bensa, M., Capo-Chichi, J.M., Delemer, B., Balsalobre, A., Nassif, C., Papadimitriou, D.T., Pagnier, A., Hasselmann, C., Patry, L., Schwartzentruber, J., Souchon, P.F., Takayasu, S., Enjalbert, A., Van Vliet, G., Majewski, J., Drouin, J., and Samuels, M.E. 2014. Mutations in NFKB2 and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies. BMC Med. Genet. 15:139. PMID: 25524009. doi: 10.1186/s12881-014-0139-9.
Chen, K., Coonrod, E.M., Kumánovics, A., Franks, Z.F., Durtschi, J.D., Margraf, R.L., Wu, W., Heikal, N.M., Augustine, N.H., Ridge, P.G., Hill, H.R., Jorde, L.B., Weyrich, A.S., Zimmerman, G.A., Gundlapalli, A.V., Bohnsack, J.F., and Voelkerding, K.V. 2013. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am. J. Hum. Genet. 93(5):812–824. PMID: 24140114. doi: 10.1016/j.ajhg.2013.09.009.
Cildir, G., Low, K.C., and Tergaonkar, V. 2016. Noncanonical NF-κB signaling in health and disease. Trends Mol. Med. 22(5):414–429. PMID: 27068135. doi: 10.1016/j.molmed.2016.03.002.
Hoeger, B., Serwas, N.K., and Boztug, K. 2018. Human NF-κB1 haploinsufficiency and Epstein–Barr virus-induced disease—Molecular mechanisms and consequences. Front. Immunol. 8:1978. PMID: 29403474. doi: 10.3389/fimmu.2017.01978.
Kuehn, H.S., Niemela, J.E., Sreedhara, K., Stoddard, J.L., Grossman, J., Wysocki, C.A., de la Morena, M.T., Garofalo, M., Inlora, J., Snyder, M.P., Lewis, D.B., Stratakis, C.A., Fleisher, T.A., and Rosenzweig, S.D. 2017. Novel nonsense gain-of-function NFKB2 mutations associated with a combined immunodeficiency phenotype. Blood. 130(13):1553–1564. PMID: 28778864. doi: 10.1182/blood-2017-05-782177.
Lal, R.A., Bachrach, L.K., Hoffman, A.R., Inlora, J., Rego, S., Snyder, M.P., and Lewis, D.B. 2017. A case report of hypoglycemia and hypogammaglobulinemia: DAVID syndrome in a patient with a novel NFKB2 mutation. J. Clin. Endocrinol. Metab. 102(7):2127–2130. PMID: 28472507. doi: 10.1210/jc.2017-00341.
Lee, C.E., Fulcher, D.A., Whittle, B., Chand, R., Fewings, N., Field, M., Andrews, D., Goodnow, C.C., and Cook, M.C. 2014. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood. 124(19):2964–2972. PMID: 25237204. doi: 10.1182/blood-2014-06-578542.
Lindsley, A.W., Qian, Y., Valencia, C.A., Shah, K., Zhang, K., and Assa’ad, A. 2014. Combined immune deficiency in a patient with a novel NFKB2 mutation. J. Clin. Immunol. 34(8):910–915. PMID: 25205549. doi: 10.1007/s10875-014-0095-3.
Liu, Y., Hanson, S., Gurugama, P., Jones, A., Clark, B., and Ibrahim, M.A. 2014. Novel NFKB2 mutation in early-onset CVID. J. Clin. Immunol. 34(6):686–690. PMID: 24888602. doi: 10.1007/s10875-014-0064-x.
Oliveira, J.B., Bleesing, J.J., Dianzani, U., Fleisher, T.A., Jaffe, E.S., Lenardo, M.J., Rieux-Laucat, F., Siegel, R.M., Su, H.C., Teachey, D.T., and Rao, V.K. 2010. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood. 116(14):e35–e40. PMID: 20538792. doi: 10.1182/blood-2010-04-280347.
Shah, S., Wu, E., Rao, V.K., and Tarrant, T.K. 2014. Autoimmune lymphoproliferative syndrome: An update and review of the literature. Curr. Allergy Asthma Rep. 14(9):462. PMID: 25086580. doi: 10.1007/s11882-014-0462-4.
Shi, C., Wang, F., Tong, A., Zhang, X.Q., Song, H.M., Liu, Z.Y., Lyu, W., Liu, Y.H., and Xia, W.B. 2016. NFKB2 mutation in common variable immunodeficiency and isolated adrenocorticotropic hormone deficiency: A case report and review of literature. Medicine. 95(40):e5081. PMID: 27749582. doi: 10.1097/MD.0000000000005081.
Sun, S.C. 2012. The noncanonical NF-κB pathway. Immunol. Rev. 246(1):125–140. PMID: 22435551. doi: 10.1111/j.1600-065X.2011.01088.x.
Turpin, D., Furudoi, A., Parrens, M., Blanco, P., Viallard, J.F., and Duluc, D. 2018. Increase of follicular helper T cells skewed toward a Th1 profile in CVID patients with non-infectious clinical complications. Clin. Immunol. 197:130–138. PMID: 30219667. doi: 10.1016/j.clim.2018.09.006.
Xiao, G., Harhaj, E.W., and Sun, S.C. 2001. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell. 7(2):401–409. PMID: 11239468. doi: 10.1016/s1097-2765(01)00187-3.
Positive newborn screen: A case of a novel variant in DCLRE1C in a patient with SCID
Noreen Choea, Lauren Brickb, Mariya Kozenkob, Pranesh Chakrobortyc, Kristin D. Kernohanc, Dennis Bulmanc, Rae Bragera
aDivision of Rheumatology, Clinical Immunology, and Allergy, Department of Pediatrics, McMaster Children’s Hospital. Hamilton, ON, Canada
bDivision of Metabolics and Genetics, Department of Pediatrics, McMaster Children’s Hospital. Hamilton, ON, Canada
cDivision of Metabolics and Newborn Screening, Department of Pediatrics, University of Ottawa, and Newborn Screening Ontario, Ottawa, ON, Canada
Background: Artemis enzyme, encoded by the DCLRE1C gene, is essential to V(D)J recombination in both T and B lymphocytes. Artemis functions as an important component of the nonhomologous end-joining DNA double-strand break repair pathway. Artemis Deficiency leads to a T-B-NK+ severe combined immune deficiency (SCID) associated with radiosensitivity.
Clinical Presentation: We present a case of a positive newborn screen for SCID in a patient who was subsequently shown to have a T-B-NK+ phenotype. Further immune evaluation showed profound T and B lymphopenia, near-absent response to mitogen stimulation, and absent immunoglobulins A and M. Genetic investigation demonstrated a novel and putative pathogenic variant in the DCLRE1C gene.
Conclusion: This case identifies a novel variant in the DCLRE1C gene in a patient with SCID identified by newborn screening.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 6 • Number 4 • December 2019
Pages: 148 - 163
Editor: Chaim M. Roifman
History
Version of record online: 17 December 2019
Copyright
© 2019.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Chaim M.Roifman. 2019. Abstracts from the Immunodeficiency Canada—7th SCID Symposium, Montreal, QC, 24 October 2019. LymphoSign Journal.
6(4): 148-163. https://doi.org/10.14785/lymphosign-2019-0015
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Management of newborn screening for severe combined immunodeficiency at a quaternary referral centre—an updated algorithm
2. Report of the Canadian Expert Committee on the management of ADA deficiency
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
