Cartilage hair hypoplasia: heterogeneity in clinical features and management among siblings
Abstract
Background: Cartilage-hair hypoplasia (CHH), caused by mutations in the ribonuclease mitochondrial RNA-processing (RMRP) gene, is associated with diverse immune abnormalities including combined immune deficiency (CID). Most patients with CHH are managed with supportive measurements, while few have received allogeneic hematopoietic stem cell transplantations (HSCT). The progression of the immune abnormalities and the impact of HSCT in patients with CHH and CID have not been well characterized.
Methods: The clinical and laboratory findings of 2 siblings diagnosed in infancy with CHH and CID due to the common 70A>G mutation in RMRP, including the effects of HSCT performed in 1 of them, were compared.
Results: Both patients suffered from recurrent respiratory infections at early age with reduced T cells numbers and responses. Patient 1 immune function continued to deteriorate leading to HSCT from an HLA-matched sibling at 4.5 years of age. The patient suffered acute and chronic graft versus host disease of the skin with residual mild joint contractures and scleroderma-like skin changes. Seven years after HSCT patient 1 has normal immune function. Immune evaluations of patient 2 in the first years of life indicated mild improvement. The patient did not have a suitable related HSCT donor and the family elected to continue with supportive care. At 7 years of age, patient 2 is clinically well and thriving with persistent T cell abnormalities.
Conclusions: Close monitoring of immune function in early life for patients with CHH and CID as well as the availability of suitable donors assists in determining management, including HSCT.
Statement of novelty: The manuscript demonstrates the importance of close monitoring and personalized approach in the management of patients affected by CHH.
Introduction
Cartilage hair hypoplasia (CHH) was initially described in the Amish and Finnish populations as a constellation of short stature, metaphyseal chondrodysplasia, and fair hypoplastic hair (McKusick et al. 1965). Subsequently, patients were also found to suffer from hypotrichosis, bone marrow dysplasia, macrocytic anemia, and hirschprung disease (Thiel and Rauch 2011). CHH is caused by autosomal recessive mutations in the non-coding ribonuclease mitochondrial RNA processing (RMRP) gene, which is involved in ribosomal assembly and cyclin-dependent cell cycle regulation (Ridanpää et al. 2001). The 70A>G mutation in the RMRP gene was previously reported to account for 92% of the Finnish CHH and 48% of the non-Finnish patients (Ridanpää et al. 2002), although many additional mutations have been identified. These mutations include insertions and duplications upstream of the transcription initiation site as well as 1–2 nucleotide changes in highly conserved residues (Thiel et al. 2007). Rapidly dividing cells such as chondrocytes and lymphocytes are particularly susceptible to RMRP defects (Thiel et al. 2005), explaining the frequent immune abnormalities observed among patients with CHH.
Autoantibodies, autoimmunity, granulomatous manifestations, and malignancies have commonly been reported in patients with CHH (Taskinen et al. 2008; Moshous et al. 2011; Biggs et al. 2017). Although most patients with CHH remain infection-free, as many as 73% display impaired humoral and or cellular immunity that may lead to recurrent or opportunistic infections including varicella zoster (Notarangelo et al. 2008; Rider et al. 2009; Kostjukovits et al. 2017c). Few patients with CHH have been identified with profound T cell deficits, resulting in a phenotype ranging from severe combined immune deficiency (SCID) (Buckley et al. 1997; Kavadas et al. 2008), Omenn’s syndrome (Roifman et al. 2006) to the less severe form of combined immune deficiency (CID) (McCann et al. 2014). Among 7 patients with CHH and profound T cell deficiency previously reported in our center, 1 had SCID features while 6 had CID, defined by reduced number of T cells (yet more than 500 CD3+ T cells/μL) and <50% stimulation index (SI) to phytohemagglutinins (PHA) (Roifman et al. 2012). Yet, the progression of the immune abnormalities in patients with CHH and CID has not been well characterized.
The diversity in the immune dysfunction among patients with CHH has led to different management strategies. Most patients require no treatment, while those with recurrent bacterial infections or with defects in antibody production to polysaccharide antigens such as pneumococci may benefit from antibiotic or immunoglobulin prophylaxis (Kostjukovits et al. 2017b). Allogeneic hematopoietic stem cell transplantations (HSCT) have been reserved for limited number of patients with CHH and severe immune abnormalities (Berthet et al. 1996; Buckley et al. 1999; Kuijpers et al. 2003; Guggenheim et al. 2006). Complications such as graft versus host disease (GvHD) and increased mortality have commonly been reported after HSCT for CHH, particularly when unrelated donors or haplo-identical parents are used (Nahum et al. 2009; Bordon et al. 2010; Ip et al. 2015; Heimall et al. 2017). Hence, the benefits of HSCT and particularly its ability to alter immune abnormalities in patients with CHH and CID require further clarifications.
Here the clinical course and immune evaluations of 2 siblings identified in infancy as suffering from CHH and CID are compared. Differences in development of immune abnormalities with age were noted, emphasizing the importance of repeated monitoring of patients with CHH and CID. Moreover, the ability of HSCT to correct an otherwise profound and persistent immune deficiency is demonstrated, further supporting the early use of HSCT for patients with persistently profound T cell abnormalities.
Patients’ descriptions
Patient 1 was born to non-consanguineous parents of French–Irish and Native American origin. At 30 weeks of gestation, an ultrasound revealed small long bones and a bell-shaped chest. The male patient was born at term with a weight of 3.4 kg. The patient was noted to have bowed legs, fine sparse hair, and constipation with no evidence of Hirschsprung disease. Genetic analysis demonstrated homozygosity for the common 70A>G mutation in the RMRP gene. The patient suffered from recurrent respiratory infections that were often treated with antibiotics. Immune assessment, first performed at 2 years of age, demonstrated appropriate immunoglobulin levels and production of antibodies to tetanus vaccine (1.38 IU/mL) and EBV. However the number of T cells and the response to PHA were reduced (Table 1). Follow-up evaluations indicated further deterioration in T cell numbers and responses. By 4 years of age, analysis of the T cell repertoire, performed by flow cytometry as previously described (Grunebaum et al. 2006), demonstrated impaired diversity of CD4+ T cells (not shown) and CD8+ T cells (Figure 1A), with expansion of some families and underrepresentation of others. An HLA-identical sibling was identified and following extensive discussions with the family, the patient underwent HSCT at 4.5 years of age. HSCT was performed with busulfan (16 mg/kg) and cyclophosphamide (200 mg/kg) conditioning as well as prednisone and cyclosporine for GvHD prophylaxis, as previously described (Grunebaum et al. 2006). The patient suffered grade 1 acute GvHD of the skin that resolved with a short course of prednisone. One year after HSCT, the patient developed skin rash. Skin biopsy demonstrated grade II acute GvHD with some features of chronic or healing GvHD. No other features of GvHD were observed. The patient was treated with prednisone with rapid response followed by a course of physiotherapy and methotrexate that was discontinued 5 years after HSCT. The patient continued to suffer from residual mild wrists and ankles joint contractures as well as mild scleroderma-like skin changes and tightness. Repeated evaluations, as late as 7 years after HSCT, demonstrated complete donor engraftment and immune reconstitution with normalization of T cell numbers and diversity, as well as response of T cells to stimulation and production of antibodies. The patient’s height at 11 years of age (Figure 2A), plotted on growth charts for CHH (Mäkitie et al. 1992) has remained on the 15th percentile.
Table 1:
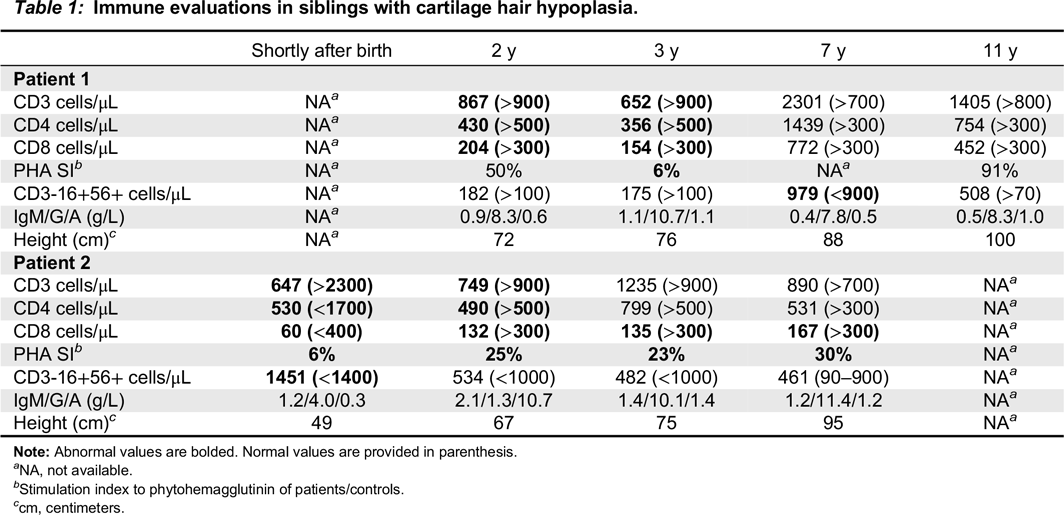
Note: Abnormal values are bolded. Normal values are provided in parenthesis.
a
NA, not available.
b
Stimulation index to phytohemagglutinin of patients/controls.
c
cm, centimeters.
Figure 1:
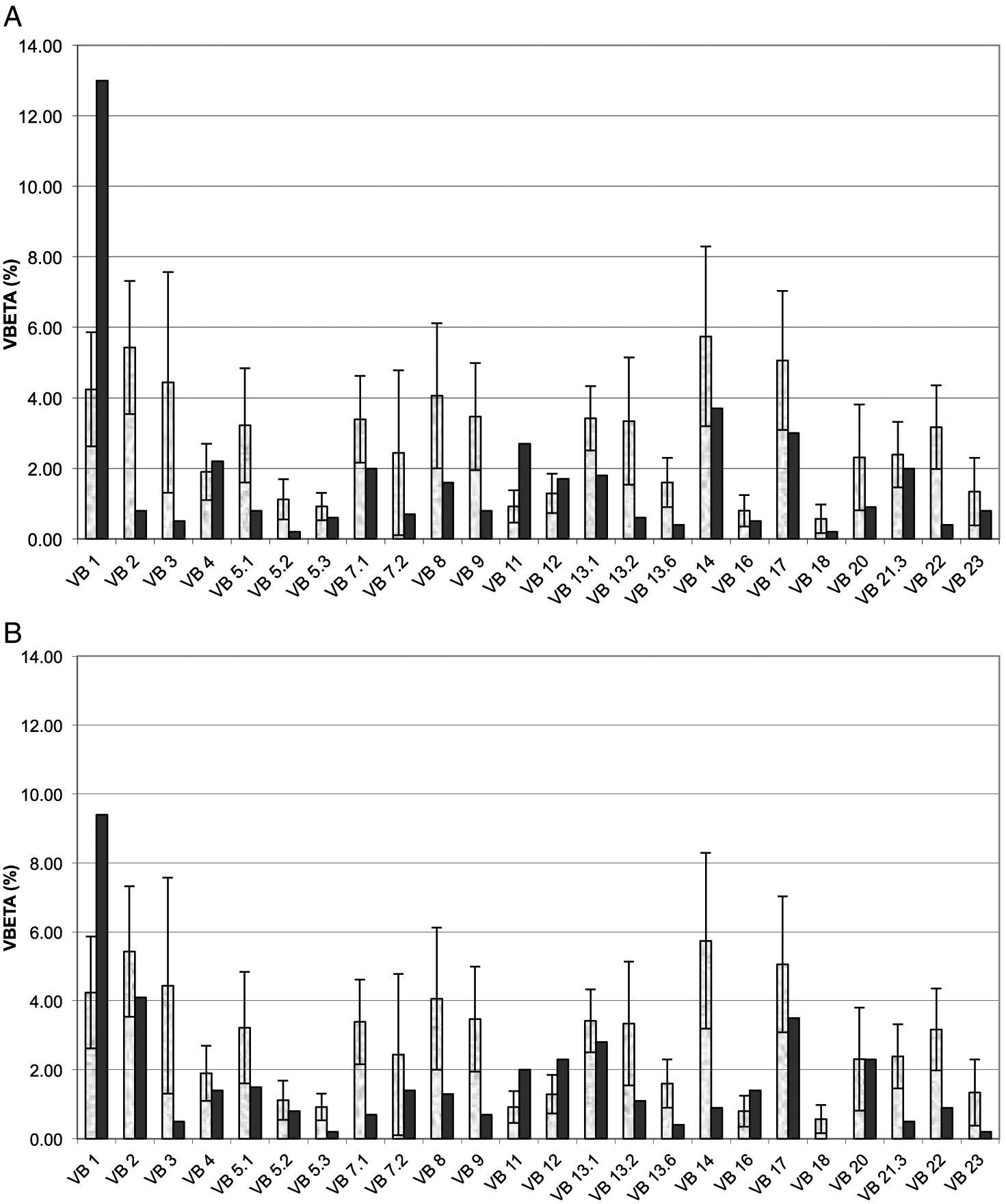
Figure 2:
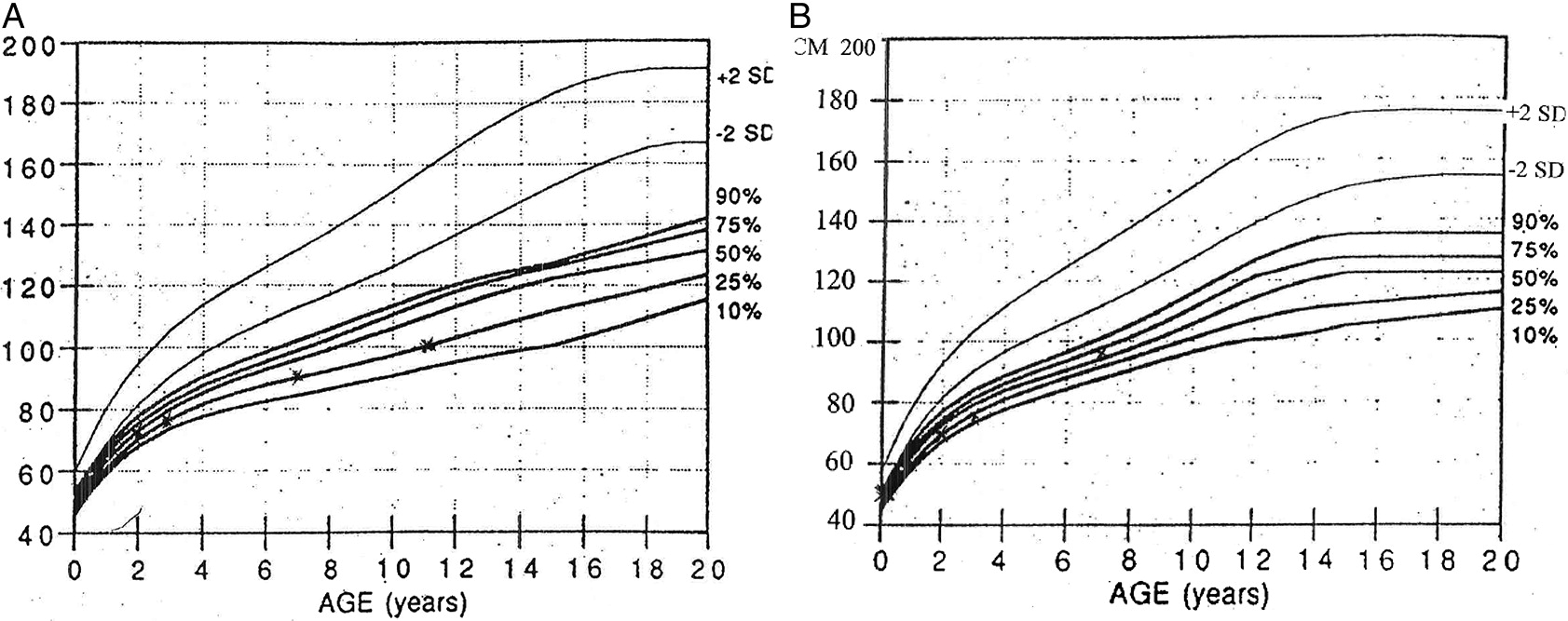
Patient 2, a female, was born 4 years after patient 1. Antenatal ultrasound showed short limbs and genetic analysis confirmed the homozygous RMRP mutations. At 10 days of age the patient suffered from respiratory syncytial virus pneumonia, requiring a 2-week admission for supportive care. In infancy, the patient had recurrent respiratory infections and constipation. Immune evaluations at 2 months of age showed T cell lymphopenia and poor T cell function (Table 1). In addition, T cell receptor excision circles, determined as previously described (Roifman et al. 2012), were moderately reduced at 220 (normal > 400/0.5 μg DNA). Pneumocystis jirovecii pneumonia prophylaxis was initiated. Subsequent laboratory evaluations demonstrated appropriate immunoglobulin levels as well as adequate production of antibodies to tetanus (4 IU/mL) and pneumococci (total IgG >270 mg/L) following vaccination. In addition, the patient had intermittent neutropenia of undetermined etiology that eventually resolved. By 3 years of age the patient’s number of CD4+ T cells improved and T cell responses stabilized at 25% ± 5% of normal. The patient did not have an HLA-matched family donor and the parents elected not to pursue HSCT using alternative donors. At 7 years of age the patient had remained clinically well with no recurrent or severe infections or autoimmunity, with improved height (Figure 2B). However, immune evaluations show persistent low number of CD8+ T cells (Table 1) with restricted repertoire (Figure 1B), reduced CD45Ra naïve CD4+ T cells (5.4% versus 43% in control) and CD8 T cells (9.6% versus 15.8%) as well as depressed T cell responses.
Appropriate standards for human experimentation were followed and informed consent was obtained prior to participating in the study.
Discussion
Improved awareness and the introduction of newborn screening for SCID have led to increased numbers of patients with CHH who are identified with profound T cell abnormalities (Kwan et al. 2013). For patients with CHH suffering from SCID or Omenn’s syndrome, HSCT is clearly indicated. However, for those with CID, disease progression and the role of HSCT have not been well characterized.
Clinical features such as extreme short stature at birth, opportunistic infections or severe autoimmunity have been proposed as predictors of severe immune dysfunction in patients with CHH (Rider et al. 2009). However, as demonstrated here, CID can still occur with near normal birth length and without severe infections. There have also been suggestions that markedly reduced IgG and absent IgA, or elevated NK cells signify a more profound immune defect (Rider et al. 2009), yet these were also not observed in the patients described here. Correlation between specific RMRP gene mutations or their locations with clinical manifestations have been investigated previously (Kostjukovits et al. 2017a). Mutations involving the promoter region have often been considered as more damaging (Notarangelo et al. 2008). Nevertheless, extensive studies of the RMRP gene have not been able to establish clear association with the severity of patients’ immune phenotype (Thiel et al. 2007; Kostjukovits et al. 2017c).
Due to the lack of predictive criteria, the patients described here were evaluated repeatedly in the first few years of life. Both siblings initially suffered from similar clinical and laboratory findings, however, subsequent assessments revealed discrepant course. Patient 1 demonstrated deteriorating immunity while the other exhibited mild improvement and stabilization of immune function. Variability between siblings with CHH has been described (Mäkitie et al. 1992; Mäkitie and Kaitila 1993; Kavadas et al. 2008). Moreover, diverse immune abnormalities have been found among patients carrying the homozygous 70A>G RMRP mutation, which was also the cause of the CHH in the siblings reported here. Among the 25 Amish and 53 Finnish patients, most with homozygous 70A>G RMRP mutations, 8% and 27%, respectively, were characterized as suffering from CID (Rider et al. 2009; Kostjukovits et al. 2017c). Whether the variability in immune dysfunction is due to unrecognized genetic or epigenetic modifying factors or exposure of different environmental triggers, such as infections, remains to be established. Nevertheless, the differences in disease progression between the 2 patients reported here emphasize the need for close monitoring of patients with CHH and CID for determination of best management.
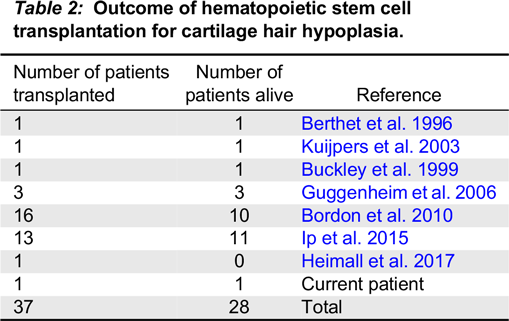
Patient 1, with deteriorating immune function and the availability of an HLA-identical sibling, received HSCT at 4.5 years of age. HSCT have been reported for 37 patients with CHH, including the patient described here, with 28 (75.6%) survivors (Table 2). Among the 6 patients who received HSCT for CHH due to homozygous 70A>G mutations (Rider et al. 2009; Bordon et al. 2010; Ip et al. 2015) 1 had died, highlighting the risks associated with HSCT. Detailed evaluations of patient 1 confirmed the success of the HSCT in normalizing immune function, an accomplishment that is further emphasized when comparing the persistent immune abnormalities observed in the non-transplanted sibling. Although patient 2 has been clinically well, her immune evaluations are ominous and indicate an increased risk of opportunistic infections, autoimmunity, and malignancy, which may adversely affect outcome of future HSCT.
Table 2:
In conclusion, the patients presented here demonstrate the variability in manifestations among patients with CHH, and the importance of close monitoring of immune function in determining best management for individual patients.
Abbreviations
- CHH
- cartilage-hair hypoplasia
- CID
- combined immunodeficiency
- GVHD
- graft versus host disease
- HSCT
- hematopoietic stem cell transplantation
- RMRP
- ribonuclease mitochondrial RNA-processing
- SCID
- severe combined immunodeficiency
REFERENCES
Berthet F., Siegrist C.A., Ozsahin H., Tuchschmid P., Eich G., Superti-Furga A., and Seger R.A. 1996. Bone marrow transplantation in cartilage-hair hypoplasia: Correction of the immunodeficiency but not of the chondrodysplasia. Eur. J. Pediatr. 155(4):286–290.
Biggs C.M., Kostjukovits S., Dobbs K., Laakso S., Klemetti P., Valta H., Taskinen M., Mäkitie O., and Notarangelo L.D. 2017. Diverse autoantibody reactivity in cartilage-hair hypoplasia. J. Clin. Immunol. 37(6):508–510.
Bordon V., Gennery A.R., Slatter M.A., Vandecruys E., Laureys G., Veys P., Qasim W., Friedrich W., Wulfraat N.M., Scherer F., Cant A.J., Fischer A., Cavazzana-Calvo M., Bredius R.G., Notarangelo L.D., Mazzolari E., Neven B., and Güngör T. 2010. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation. Blood. 116(1):27–35.
Buckley R.H., Schiff R.I., Schiff S.E., Markert M.L., Williams L.W., Harville T.O., Roberts J.L., and Puck J.M. 1997. Human severe combined immunodeficiency: Genetic, phenotypic, and functional diversity in one hundred eight infants. J. Pediatr. 130(3):378–387.
Buckley R.H., Schiff S.E., Schiff R.I., Markert M.L., Williams L.W., Roberts J.L., Myers L.A., and Ward F.E. 1999. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N. Engl. J. Med. 340(7):508–516.
Grunebaum E., Mazzolari E., Porta F., Dallera D., Atkinson A., Reid B., Notarangelo L.D., and Roifman C.M. 2006. Bone marrow transplantation for severe combined immune deficiency. JAMA. 295(5):508–518.
Guggenheim R., Somech R., Grunebaum E., Atkinson A., and Roifman C.M. 2006. Bone marrow transplantation for cartilage-hair-hypoplasia. Bone Marrow Transplant. 38(11):751–756.
Heimall J., Logan B.R., Cowan M.J., Notarangelo L.D., Griffith L.M., Puck J.M., Kohn D.B., Pulsipher M.A., Parikh S., Martinez C., Kapoor N., O’Reilly R., Boyer M., Pai S.Y., Goldman F., Burroughs L., Chandra S., Kletzel M., Thakar M., Connelly J., Cuvelier G., Davila B., Shereck E., Knutsen A., Sullivan K.E., DeSantes K., Gillio A., Haddad E., Petrovic A., Quigg T., Smith A.R., Stenger E., Yin Z., Shearer W.T., Fleisher T., Buckley R.H., and Dvorak C.C. 2017. Immune reconstitution and survival of 100 SCID patients post hematopoietic cell transplant: A PIDTC natural history study. Blood. 130(25):2718–2727.
Ip W., Gaspar H.B., Kleta R., Chanudet E., Bacchelli C., Pitts A., Nademi Z., Davies E.G., Slatter M.A., Amrolia P., Rao K., Veys P., Gennery A.R., and Qasim W. 2015. Variable phenotype of severe immunodeficiencies associated with RMRP gene mutations. J. Clin. Immunol. 35(2):147–157.
Kavadas F.D., Giliani S., Gu Y., Mazzolari E., Bates A., Pegoiani E., Roifman C.M., and Notarangelo L.D. 2008. Variability of clinical and laboratory features among patients with ribonuclease mitochondrial RNA processing endoribonuclease gene mutations. J. Allergy Clin. Immunol. 122(6):1178–1184.
Kostjukovits S., Degerman S., Pekkinen M., Klemetti P., Landfors M., Roos G., Taskinen M., and Mäkitie O. 2017a. Decreased telomere length in children with cartilage-hair hypoplasia. J. Med. Genet. 54(5):365–370.
Kostjukovits S., Klemetti P., Föhr A., Kajosaari M., Valta H., Taskinen M., Toiviainen-Salo S., and Mäkitie O. 2017b. High prevalence of bronchiectasis in patients with cartilage-hair hypoplasia. J. Allergy Clin. Immunol. 139(1):375–378.
Kostjukovits S., Klemetti P., Valta H., Martelius T., Notarangelo L.D., Seppänen M., Taskinen M., and Mäkitie O. 2017c. Analysis of clinical and immunologic phenotype in a large cohort of children and adults with cartilage-hair hypoplasia. J. Allergy Clin. Immunol. 140(2):612–614.e5.
Kuijpers T.W., Ridanpää M., Peters M., de Boer I., Vossen J.M.J.J., Pals S.T., Kaitila I., and Hennekam R.C. 2003. Short-limbed dwarfism with bowing, combined immune deficiency, and late onset aplastic anaemia caused by novel mutations in the RMPR gene. J. Med. Genet. 40:761–766.
Kwan A., Church J.A., Cowan M.J., Agarwal R., Kapoor N., Kohn D.B., Lewis D.B., McGhee S.A., Moore T.B., Stiehm E.R., Porteus M., Aznar C.P., Currier R., Lorey F., and Puck J.M. 2013. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: Results of the first 2 years. J. Allergy Clin. Immunol. 132(1):140–150.e7.
Mäkitie O. and Kaitila I. 1993. Cartilage-hair hypoplasia—Clinical manifestations in 108 Finnish patients. Eur. J. Pediatr. 152(3):211–217.
Mäkitie O., Perheentupa J., and Kaitila I. 1992. Growth in cartilage-hair hypoplasia. Pediatr. Res. 31(2):176–180.
McCann L.J., McPartland J., Barge D., Strain L., Bourn D., Calonje E., Verbov J., Riordan A., Kokai G., Bacon C.M., Wright M., and Abinun M. 2014. Phenotypic variations of cartilage hair hypoplasia: Granulomatous skin inflammation and severe T cell immunodeficiency as initial clinical presentation in otherwise well child with short stature. J. Clin. Immunol. 34(1):42–48.
McKusick V.A., Eldridge R., Hostetler J.A., Ruangwit U., and Egeland J.A. 1965. Dwarfism in the Amish. II. Cartilage-hair hypoplasia. Bull. Johns Hopkins Hosp. 116:285–326.
Moshous D., Meyts I., Fraitag S., Janssen C.E., Debré M., Suarez F., Toelen J., De Boeck K., Roskams T., Deschildre A., Picard C., Bodemer C., Wouters C., and Fischer A. 2011. Granulomatous inflammation in cartilage-hair hypoplasia: Risks and benefits of anti-TNF-α mAbs. J. Allergy Clin. Immunol. 128(4):847–853.
Nahum A., Reid B., Grunebaum E., and Roifman C.M. 2009. Matched unrelated bone marrow transplant for Omenn syndrome. Immunol. Res. 44(1–3):25–34.
Notarangelo L.D., Roifman C.M., and Giliani S. 2008. Cartilage-hair hypoplasia: Molecular basis and heterogeneity of the immunological phenotype. Curr. Opin. Allergy Clin. Immunol. 8(6):534–539.
Ridanpää M., Sistonen P., Rockas S., Rimoin D.L., Kakitie O., and Kaitila I. 2002. Worldwide mutation spectrum in cartilage-hair hypoplasia: Ancient founder origin of the major 70A→G mutation of the untranslated RMRP. Eur. J. Hum. Genet. 10:439–447.
Ridanpää M., van Eenennaam H., Pelin K., Chadwick R., Johnson C., Yuan B., van Venrooij W., Pruijn G., Salmela R., Rockas S., Mäkitie O., Kaitila I., and de la Chapelle A. 2001. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell. 104(2):195–203.
Rider N.L., Morton D.H., Puffenberger E., Hendrickson C.L., Robinson D.L., and Strauss K.A. 2009. Immunologic and clinical features of 25 Amish patients with RMRP 70 A → G cartilage hair hypoplasia. Clin. Immunol. 131(1):119–128.
Roifman C.M., Gu Y., and Cohen A. 2006. Mutations in the RNA component of RNase mitochondrial RNA processing might cause Omenn syndrome. J. Allergy Clin. Immunol. 117(4):897–903.
Roifman C.M., Somech R., Kavadas F., Pires L., Nahum A., Dalal I., and Grunebaum E. 2012. Defining combined immunodeficiency. J. Allergy Clin. Immunol. 130(1):177–183.
Taskinen M., Ranki A., Pukkala E., Jeskanen L., Kaitila I., and Mäkitie O. 2008. Extended follow-up of the Finnish cartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am. J. Med. Genet. A. 146A(18):2370–2375.
Thiel C.T. and Rauch A. 2011. The molecular basis of the cartilage-hair hypoplasia–anauxetic dysplasia spectrum. Best Pract. Res. Clin. Endocrinol. Metab. 25(1):131–142.
Thiel C.T., Horn D., Zabel B., Ekici A.B., Salinas K., Gebhart E., Rüschendorf F., Sticht H., Spranger J., Müller D., Zweier C., Schmitt M.E., Reis A., and Rauch A. 2005. Severely incapacitating mutations in patients with extreme short stature identify RNA-processing endoribonuclease RMRP as an essential cell growth regulator. Am. J. Hum. Genet. 77(5):795–806.
Thiel C.T., Mortier G., Kaitila I., Reis A., and Rauch A. 2007. Type and level of RMRP functional impairment predicts phenotype in the cartilage hair hypoplasia–anauxetic dysplasia spectrum. Am. J. Hum. Genet. 81(3):519–529.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 5 • Number 1 • March 2018
Pages: 21 - 28
History
Received: 12 December 2017
Accepted: 9 January 2018
Accepted manuscript online: 10 January 2018
Copyright
© 2018.
Authors
Funding Information
EG is supported in part by the Donald and Audrey Campbell Chair in Immunology.
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AmiirahAujnarain, Vy Hong-DiepKim, and EyalGrunebaum. 2018. Cartilage hair hypoplasia: heterogeneity in clinical features and management among siblings. LymphoSign Journal.
5(1): 21-28. https://doi.org/10.14785/lymphosign-2017-0014
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
