Novel mutations in the RNU4ATAC gene define obligatory domain changes for Roifman syndrome
Yael Dinur Schejtera, Brenda Reida, Daniele Mericob, Chaim M. Roifmana,c
aDivision of Immunology and Allergy, Department of Pediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada; bDeep Genomics, University of Toronto Early-Stage Technology, University of Toronto, Toronto, ON, Canada; cThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children, Toronto, ON, Canada
Introduction: Roifman syndrome (OMIM #616651) is characterized by remarkably consistent features of humoral immunodeficiency, pre- and postnatal growth retardation, spondyloepiphyseal dysplasia, developmental delay, retinal dystrophy, and unique facial dysmorphism (Roifman 1999). Other features have been inconsistently described including cardiac noncompaction (Mandel et al. 2001), hypogonadotrophic hypogonadism (Robertson et al. 2000), partial agenesis of the corpus callosum, hypocampal hypoplasia (Fairchild et al. 2011), and renal tubular dysfunction (Fairchild et al. 2011). Recently, rare compound heterozygote mutations in the RNU4ATAC gene were found to be responsible for this disorder (Merico et al. 2015).
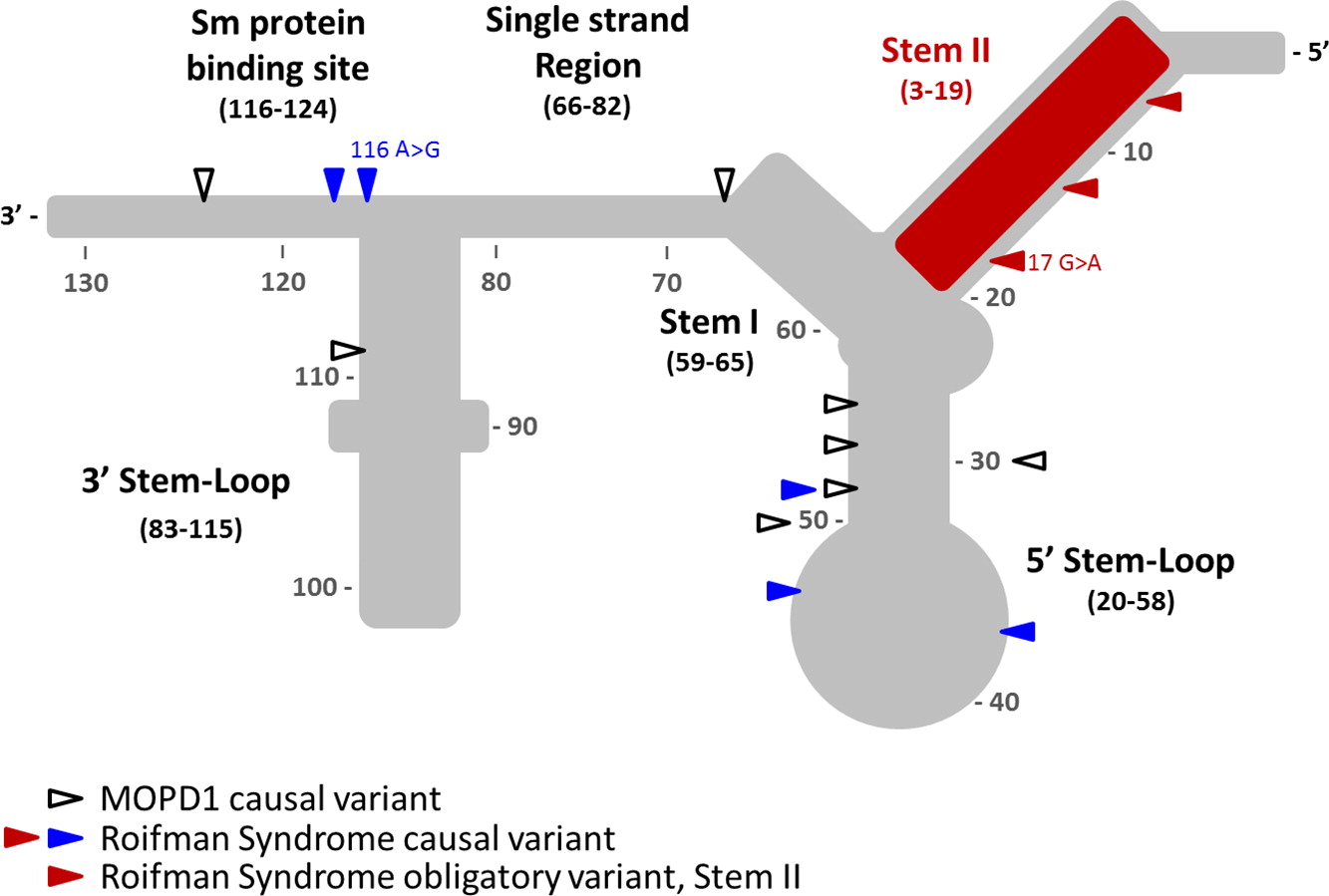
RNU4ATAC encodes for U4atac small nuclear RNA (snRNA), an essential component of the minor spliceosome, which is crucial for the correct splicing of about 800 genes carrying minor introns (Tarn and Steitz 1996). The structural elements of U4atac snRNA include stem I and stem II, which base pair with U6atac to form the catalytically active minor spliceosome, and are separated by a 5′ stem-loop. A 3′ stem-loop connects the binding site for Sm proteins, which is required for the assembly of the complex and for import into the nucleus. It remains unclear which domains control splicing of some or all of the 800 different genes.
To date, Roifman syndrome causal variants present with a characteristic compound heterozygosity pattern, with one variant involving the 5′ stem-loop or the Sm protein-binding site, and the other variant affecting the stem II element—a newly implicated, yet highly conserved, element of the gene. In contrast, homozygous or compound heterozygous mutations in the 5′ stem-loop (position 20–58) and the Sm protein binding site are associated with a completely different and clinically distinctive entity, MOPD1 (microcephalic osteodyslastic primordial dwarfism type I). MOPD1 is associated with pre- and postnatal lethality, severe prenatal microcephaly and brain malformations, intractable epilepsy, short and bowed limbs, absent or sparse hair, neuroendocrine dysfunction, and distinct facial features including proptotic eyes, prominent nose or downturned nasal tip, micrognathia, and occasionally, skin and retinal hypopigmentation.
Together, these different and distinct clinical syndromes suggest diverse functions are assigned to the various domains of U4atac small nuclear RNA.
Methods and Results:
Case description: The patient is a 12-year-old boy of Tamil descent, born at term with a low birth weight of 1.8 kg. He first presented at 11 weeks of age with a severe bilateral pneumonia and a complicated course requiring intubation and high-flow ventilation. He then continued to suffer from recurrent pneumonia as well as a reactive airway disease.
In addition, the patient has global developmental delay. A brain MRI revealed mild ventriculomegaly and prominent extra-axial CSF (cerebrospinal fluid) spaces. He had dysmorphic features, including short stature, brachydactyly, microcephaly and hypoplastic shallow mid facies with a long philtrum and a thin upper lip. A skeletal survey at 6.5 years of age revealed diffuse spondylo-epiphyseal dysplasia.
At the age of 7 years, he was found to have complex retinal dystrophy with cystoid macular edema. He improved clinically on Brinzoamide drops.
The parents are nonconsanguineous and there is no history of primary immune deficiency in the family. The patient has an older, healthy sister and the mother has a history of 4 miscarriages.
An initial immunologic evaluation was significant for lymphopenia (1.53 × 109 cells/L) with normal eosinophil count. He had significantly reduced CD19 cell count (47 cells/µL). Numbers of other lymphocyte markers were normal (CD3 count of 3417 cells/µL, CD4 count of 2604 cells/µL, CD8 count of 837 cells/µL, and NK cell count of 387 cells/µL). He had low immunoglobulin levels (IgG of 1.8 g/L, IgA of less than 0.1 g/L, and IgM of 0.2 g/L). T-cell mitogen response was normal.
The patient’s immunoglobulin levels normalized over time, but he remained incapable of mounting a sustained response to tetanus and pneumococcal vaccines and had nonprotective titres for measles, mumps, rubella, and varicella, despite repeated vaccination. He was therefore started on IVIG treatment at the age of 11 years. Despite this treatment, the patient’s respiratory status continued to deteriorate and he eventually developed bronchiolitis obliterans following adenoviral infection.
Analysis of the RNU4ATAC gene revealed compound heterozygous mutation with a 116A>G mutation in 3′ stem loop domain and a 17G>A mutation in the Stem II domain (Figure 1).
Figure 1:
Figure 1: U4atac snRNA secondary structure elements. MOPD1 (open triangles) and Roifman Syndrome causal variants located on MOPD1-implicated elements (blue triangles) are shown. Roifman Syndrome causal variants (red triangles) located in the obligatory and highly conserved domain in Stem II are highlighted in red. Novel mutations identified in this report include the 116A>G (Sm protein binding site).
Discussion: We report a new patient with a novel mutation in the RNU4ATAC gene causing Roifman syndrome. Our patient presented with a similar phenotype to previously reported cases, with IUGR (intrauterine growth restriction), dysgammaglobulinemia, developmental delay, spondyloepiphyseal dysplasia, and distinctive dysmorphism including microcephaly and hypoplastic shallow mid facies with a long philtrum and a thin upper lip. As opposed to previous reports, he did not have eosinophilia, elevated IgE, or eczema; he did, however, suffer from reactive airway disease. He suffered from severe pneumonia requiring intubation at 11 weeks of age, which is unusually early for isolated humoral defects.
Prior to this report, all of the mutations described in Roifman syndrome patients are confined to 2 distinctive domains: one variant in the 5′ stem-loop (positions 20–58) or the Sm protein binding site (positions 117–124), both of which are implicated in MOPD1, and a second variant in the highly conserved domain in the stem II region (positions 3–19), which is unique to Roifman syndrome.
The mutation described in our patient is important in that it expands the previously reported Sm protein binding site. Indeed, the previously reported Sm protein binding site (Shukla et al. 2002) was recently revised to include position 116 (Jafarifar et al. 2014).
Conclusion: We report a new case with Roifman Syndrome that adds new important clinical and immunological information.
Research is currently underway in our laboratory as well as those of our collaborators to define the target genes affected and to explore novel genetic aberrations causing antibody deficiency, retinopathey, skeletal dysplasia as well as other features associated with Roifman syndrome.
REFERENCES
Fairchild, H.R., Fairchild, G., Tierney, K.M., McCartney, D.L., Cross, J.J., and de Vries, P.J. 2011. Partial agenesis of the corpus callosum, hippocampal atrophy, and stable intellectual disability associated with Roifman syndrome. Am. J. Med. Genet. A. Part A. 155A:2560–2565. PMID: 21910238. doi: https://doi.org/10.1002/ajmg.a.34215.
Gray, P.E., Sillence, D., and Kakakios, A. 2011. Is Roifman syndrome an X-linked ciliopathy with humoral immunodeficiency? Evidence from 2 new cases. Int. J. Immunogenet. 38:501–505. PMID: 21977988. doi: https://doi.org/10.1111/j.1744-313X.2011.01041.x.
Jafarifar, F., Dietrich, R.C., Hiznay, J.M., and Padgett, R.A. 2014. Biochemical defects in minor spliceosome function in the developmental disorder MOPD I. RNA. 20:1078–1089. PMID: 24865609. doi: https://doi.org/10.1261/rna.045187.114.
Mandel, K., Grunebaum, E., and Benson, L. 2001. Noncompaction of the myocardium associated with Roifman syndrome. Cardiol. Young. 11:240–243.
Merico, D., Roifman, M., Braunschweig, U., Yuen, R.K., Alexandrova, R., Bates, A., Reid, B., Nalpathamkalam, T., Wang, Z., Thiruvahindrapuram, B., Gray, P., Kakakios, A., Peake, J., Hogarth, S., Manson, D., Buncic, R., Pereira, S.L., Herbrick, J.A., Blencowe, B.J., Roifman, C.M., and Scherer, S.W. 2015. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat. Commun. 6:8718. PMID: 26522830. doi: https://doi.org/10.1038/ncomms9718.
Robertson, S.P., Rodda, C., and Bankier, A. 2000. Hypogonadotrophic hypogonadism in Roifman syndrome. Clin. Genet. 57:435–438. PMID: 10905663.
Roifman, C.M. 1999. Antibody deficiency, growth retardation, spondyloepiphyseal dysplasia and retinal dystrophy: A novel syndrome. Clin. Genet. 55:103–109. PMID: 10189087.
Shukla, G.C., Cole, A.J., Dietrich, R.C., and Padgett, R.A. 2002. Domains of human U4atac snRNA required for U12-dependent splicing in vivo. Nucleic Acids Res. 30:4650–4657. PMID: 12409455.
Tarn, W.Y., and Steitz, J.A. 1996. Highly diverged U4 and U6 small nuclear RNAs required for splicing rare AT-AC introns. Science. 273:1824–1832. PMID: 8791582.
Refractory auto-immune uveitis successfully treated with sirolimus as an autoimmune manifestation of a hemizygous FOXP3 mutation
Guilhem Cros, Roxane Labrosse, Isabel Fernandez, Francoise Le Deist, Eric Fortin, Elie Haddad
CHU Sainte-Justine, University of Montreal, Montreal, QC, Canada
IPEX (Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked) syndrome is characterized by systemic autoimmunity, and is caused by hemizygous mutations in the FOXP3 gene. The clinical spectrum has been progressively extended to a less severe phenotype with atypical autoimmunity and no clear genotype correlation.
We report a 16-year-old boy, with no family history, who presented with type 1 diabetes (T1D) at 5 months of age. He was diagnosed with bilateral uveitis at age 6, which was initially treated with topic corticosteroids, methotrexate, and azathioprine. Despite maximal treatment including adalimumab, his uveitis was poorly controlled and was complicated by steroid-induced cataracts and glaucoma. His clinical picture was free of other autoimmune manifestations.
Given the atypical severity of the uveitis, immunological testing was performed, and despite the normal expression of FOXP3 genetic analysis was performed and a hemizygous mutation of the FOXP3 gene was found (1040G>A, R347H), which has previously been described with different phenotypes such as severe enteropathy, eczema, AIHA.
A treatment with sirolimus was initiated and a complete remission was quickly obtained and steroid therapy could be stopped.
Patients with early-onset T1D combined with other autoimmune manifestations should be tested for FOXP3 mutations as it modifies the therapeutic strategies.
Interstitial lung disease in infancy: an unusual presentation of CD40 ligand deficiency
Victoria E. Cooka, Connie L. Yangb, Anna F. Leec, Kyla J. Hildebranda, Anne Junkera, Stuart E. Turveya
aDivision of Clinical Immunology and Allergy, Department of Pediatrics, University of British Columbia, Vancouver, BC, Canada; bDivision of Respirology, Department of Pediatrics, University of British Columbia, Vancouver, BC, Canada; cDepartment of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada
Background: Diffuse lung disease is rare in children, and recent classification systems recognize that certain disorders are more prevalent in infants (Vece and Young 2016). Childhood interstitial lung disease (chILD) syndrome describes a collection of respiratory symptoms and diffuse abnormalities on imaging (Kurland et al. 2013; Vece and Young 2016). Increasingly, immunodeficiency and immune dysregulation syndromes are recognized as causes of chILD (Vece and Young 2016).
Class switch recombination (CSR) defects are a major group of primary immunodeficiencies. CD40 ligand (CD40L) deficiency, also known as hyper-immunoglobulin M syndrome type 1, is the most common form of CSR syndrome (Korthäuer et al. 1993; Winkelstein et al. 2003; Davies and Thrasher 2010). CD40L deficiency is an X-linked recessive disorder caused by mutations in CD40L on chromosome Xq26 (Villa et al. 1994); the gene encodes a membrane glycoprotein that is expressed on activated CD4+ T cells. It binds CD40, expressed on B lymphocytes and antigen presenting cells (APC), leading to immunoglobulin class switching, generation of memory B cells, and APC-mediated activation of CD8+ T cells (Noelle et al. 1992; Hollenbaugh et al. 1994).
Affected patients, typically males, present in infancy and early childhood with pulmonary infections due to impaired humoral immunity, and opportunistic infections due to impaired cellular immunity. Autoimmune disorders and malignancies are also common (Davies and Thrasher 2010). Disease phenotype and severity are influenced by the specific gene mutation and degree of protein expression (Buchbinder et al. 2012; Katta et al. 2013). Investigations reveal normal to elevated IgM and low or undetectable IgG, IgA, and IgE. Responses to protein antigens are impaired; however, IgM isohemagglutinins may be present (Davies and Thrasher 2010). T-, B-, and NK-cell numbers are typically normal; however, specific T-cell responses may be impaired. Neutropenia is common.
Chronic pulmonary changes, including interstitial lung disease, have been described in patients with both primary and secondary immunodeficiency. Biopsies commonly demonstrate follicular bronchiolitis, lymphoid hyperplasia, and lymphoid interstitial pneumonia (Dishop 2011). Such findings on lung biopsy should prompt more detailed immunologic investigations. Diffuse pulmonary interstitial glycogenosis (PIG) has been described as a primary interstitial lung disease in infants (Canakis et al. 2002), but patchy PIG is increasingly recognized in association with lung growth abnormalities and cardiac disease (Deterding 2010). It has not previously been described in association with immunodeficiency. Here we describe a case of CD40L deficiency with a novel mutation, presenting in infancy with chILD syndrome and lung biopsy findings of patchy PIG and lymphoid aggregates.
Methods: Clinical data were obtained from patient history, assessment, and chart review. Informed consent was obtained. A left lingula wedge biopsy of lung was obtained and needle-inflated with formalin for tissue fixation. Fixed tissue sections were stained with routine hematoxylin and eosin, and special stains for glycogen and pathogenic organisms. Retrospective testing of the newborn blood spot for T-cell receptor excision circles (TRECs) was sent to the Ontario newborn screening program. CD40L expression on PMA (phorbol 12-myristate 13-acetate) and ionomycin-stimulated CD3+ CD8− T cells was measured with flow cytometry by Calgary Laboratory Services. Whole blood samples were collected from the patient and both parents and subjected to whole exome sequencing (WES) (GeneDx).
Results:
Clinical features: The patient was the first and only child to healthy, nonconsanguineous, Caucasian parents. He was born at 41 weeks via caesarean section for cephalopelvic disproportion. Birth weight was 4260 g. He was formula fed with good growth. Immunizations, including rotavirus, were given without incident. There is no family history of primary immunodeficiency, recurrent infections, early childhood death, autoimmunity, or malignancy.
He presented at 3 months of age following 2 weeks of poor oral intake, weight loss, and tachypnea. His weight was 6.15 kg (25–50 percentiles). He was tachypneic with mild hypoxia responsive to supplemental oxygen (0.5 L/min). Chest X-ray demonstrated hyperinflation, prominant perihilar markings, and bilateral subsegmental consolidation. Thymic tissue was present and cardiac silhouette was normal. He was managed with supplemental oxygen, nasogastric (NG) feeding, and 5 days of azithromycin. No infectious causes were identified. He was transferred to the tertiary care centre 1 week later for lack of clinical improvement.
Repeat microbiological testing was negative. A gastroenterology assessment indicated gastroesophageal reflux disease, cow’s milk protein allergy, and oral aversion. Sweat chloride testing, biopsy for primary ciliary dyskinesia, an upper GI study, and echocardiogram were all normal. Immunologic workup revealed normal IgM, undetectable IgA, and low IgG felt to be related to the nadir. Anti-tetanus and diphtheria toxoid titres were nonprotective after 2-month vaccines. Lymphocyte enumeration was normal.
He was treated with amoxicillin, omeprazole, and hypoallergenic formula via NG. He showed gradual clinical improvement, although tachypnea and mild hypoxia (88%–90%) persisted. Persistence of oral aversion necessitated ongoing NG feeds with hypoallergenic formula at discharge.
From 4–6 months of age, growth and stools normalized, but respiratory status remained unchanged. A diagnosis of chILD was considered. The patient underwent a CT scan that was felt to be concerning for interstitial lung disease. A lung biopsy found patchy PIG and scattered lymphoid aggregates in keeping with interstitial pneumonitis. No microorganisms were identified by culture or staining.
At 9 months of age, the patient developed worsening tachypnea and hypoxia. He was afebrile and otherwise well appearing. Bilateral bronchoalveolar lavage identified PJP (Pneumocystis jiroveci pneumonia), and blood PCR (polymerase chain reaction) identified CMV (Cytomegalovirus) viremia. Repeat immunologic testing showed undetectable IgG and IgA with normal IgM and absent vaccine responses. T-, B-, and NK-cell quantitation indicated no abnormalities. Mitogen stimulation studies revealed normal responses to PWM (pokeweed mitogen) and SAC (Staphylococcus aureus Cowen) but reduced T-cell response to PHA (phytohemagglutinin). Retrospective analysis of the newborn blood spot for TRECs was normal.
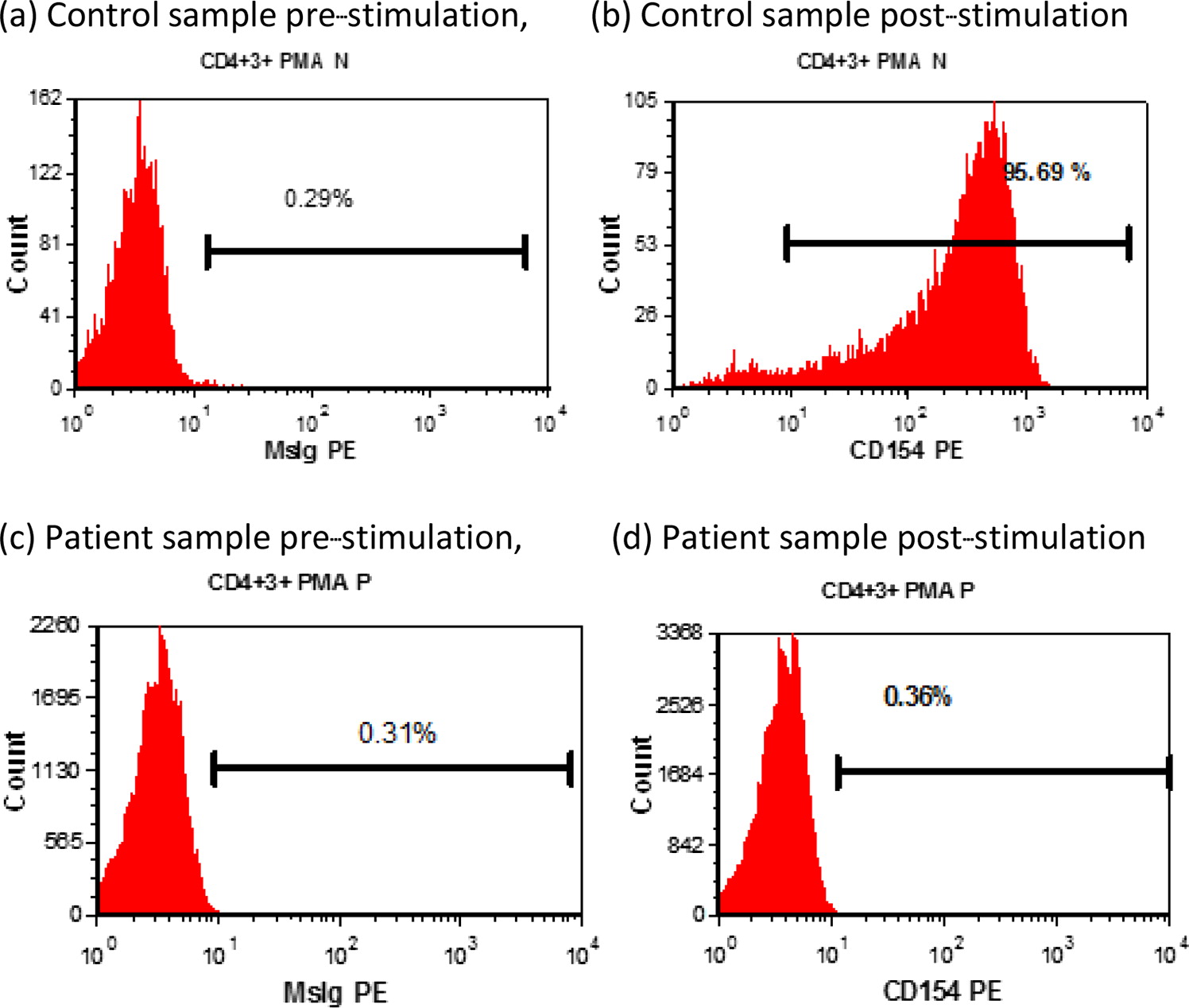
Functional studies demonstrated absent CD40L expression on activated CD4+ T cells (Figure 1), and WES revealed maternal inheritance of a novel mutation in CD40L (c.464 T>A). The patient improved clinically and is maintained on prophylactic oral trimethoprim-sulfamethoxazole and IVIG while awaiting bone marrow transplant.
Figure 1:
Figure 1: CD40L (CD154) expression by PMA and ionomycin-stimulated CD3+ CD8− T cells.
Pulmonary pathology: Lung pathology showed normal lobular architecture. The alveolar septa in some lobules were abnormally thickened due to an increased number of interstitial mesenchymal cells. A small subset of these cells contained flecks of PAS-positive, diastase-sensitive cytoplasmic glycogen, suggestive of patchy PIG. Scattered lymphoid aggregates were also seen. There was evidence of chronic bronchiolar inflammation. There was no viral cytopathic effect, and no organisms were identified with special staining. The overall impression was that of interstitial pneumonitis of unknown etiology, with a histologic differential diagnosis of lung injury versus an immunodeficiency or autoimmune disease.
Immunological features: CD40L expression by PMA and ionomycin-stimulated CD3+ CD8− T cells was low (0.36%); expression by the normal control was 95.69% (Figure 1).
Genetic analysis: WES revealed a c.464 T>A mutation in exon 5 of CD40L, resulting in an amino acid substitution, p. Leu155Gln, expected to impact secondary protein structure. Sequencing of parent samples confirmed maternal transmission.
Conclusions: This case describes a patient with CD40L deficiency presenting initially with chILD syndrome, with tachypnea, hypoxia, failure to thrive, and diffuse abnormalities on chest CT. Identification of his underlying diagnosis was delayed until he developed the more classical findings of PJP pneumonia and CMV viremia. The identified mutation, c.464 T>A in exon 5 of CD40L, is confirmed to be pathogenic based on the patient’s clinical presentation and demonstration of minimal CD40L expression by activated CD3+CD8− T cells. This mutation has not previously been reported, although CD40L deficiency secondary to a different missense variant at the same amino acid position has been described (Lin et al. 1996).
The 2013 American Thoracic Society guideline on infant interstitial lung disease suggests evaluation for immunodeficiency in patients presenting with chILD syndrome. This patient had an immune workup at the time of initial presentation; however, his minor abnormalities were felt to be maturational. This illustrates the importance of close follow-up through early childhood with repeated assessment of immunoglobulins and vaccine responses.
A recent review highlights the evolving role of WES for evaluation of diffuse lung diseases (Vece and Young 2016); recent case reports have identified genetic changes impacting CTLA4 trafficking and STAT3 signaling (Vece and Young 2016). Application of this diagnostic tool earlier in our patient’s course would also have shortened time to diagnosis.
To the best of our knowledge, this is the first published case of CD40L deficiency presenting with chILD syndrome. Our case indicates that this diagnosis should be considered in patients presenting with signs and symptoms of chILD syndrome, and it highlights the importance of thorough immunologic workup and follow-up in this patient population. Use of WES in the diagnostic workup of chILD may facilitate earlier diagnosis.
REFERENCES
Buchbinder, D., Park, S., and Nugent, D. 2012. X-linked hyper IgM syndrome: A novel sequence variant associated with an atypical mild phenotype. J. Pediatr. Hematol. Oncol. 34(5):e212–e214.
Canakis, A.M., Cutz, E., Manson, D., and O’Brodovich, H. 2002. Pulmonary interstitial glycogenosis: A new variant of neonatal interstitial lung disease. Am. J. Respir. Crit. Care Med. 165(11):1557–1565. PMID: 12045133. doi: https://doi.org/10.1164/rccm.2105139.
Hollenbaugh, D., Wu, L.H., Ochs, H.D., Nonoyama, S., Grosmaire, L.S., Ledbetter, J.A., Noelle, R.J., Hill, H., and Aruffo, A. 1994. The random inactivation of the X chromosome carrying the defective gene responsible for X-linked hyper IgM syndrome (X-HIM) in female carriers of HIGM1. J. Clin. Invest. 94(2):616–622. PMID: 7518839. doi: https://doi.org/10.1172/JCI117377.
Katta, A., Hong, J., and Knutsen, A.P. 2013. Hyper immunoglobulin M syndrome in a 15-year-old boy caused by a Gly219Arg missense mutation. Ann. Allergy Asthma Immunol. 110(5):391–393. PMID: 23622016. doi: https://doi.org/10.1016/j.anai.2013.02.011.
Korthäuer, U., Graf, D., Mages, H.W., Brière, F., Padayachee, M., Malcolm, S., Ugazio, A.G., Notarangelo, L.D., Levinsky, R.J., and Kroczek, R.A. 1993. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 361(6412):539–541. PMID: 7679206. doi: https://doi.org/10.1038/361539a0.
Kurland, G., Deterding, R.R., Hagood, J.S., Young, L.R., Brody, A.S., Castile, R.G., Dell, S., Fan, L.L., Hamvas, A., Hilman, B.C., Langston, C., Nogee, L.M., and Redding, G.J. 2013. An official American thoracic society clinical practice guideline: Classification, evaluation, and management of childhood interstitial lung disease in infancy. Am. J. Respir. Crit. Care Med. 188(3):376–394. PMID: 23905526. doi: https://doi.org/10.1164/rccm.201305-0923ST.
Lin, Q., Rohrer, J., Allen, R.C., Larché, M., Greene, J.M., Shigeoka, A.O., Gatti, R.A., Derauf, D.C., Belmont, J.W., and Conley, M.E. 1996. A single strand conformation polymorphism study of CD40 ligand. Efficient mutation analysis and carrier detection for X-linked hyper IgM syndrome. J. Clin. Invest. 97(1):196–201. PMID: 8550833. doi: https://doi.org/10.1172/JCI118389.
Noelle, R.J., Roy, M., Shepherd, D.M., Stamenkovic, I., Ledbetter, J.A., and Aruffo, A. 1992. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc. Natl. Acad. Sci. U.S.A. 89(14):6550–6554. PMID: 1378631.
Villa, A., Notarangelo, L.D., Di Santo, J.P., Macchi, P.P., Strina, D., Frattini, A., Lucchini, F., Patrosso, C.M., Giliani, S., and Mantuano, E. 1994. Organization of the human CD40L gene: Implications for molecular defects in X chromosome-linked hyper-IgM syndrome and prenatal diagnosis. Proc. Natl. Acad. Sci. U.S.A. 91(6):2110–2114. PMID: 7907793.
Winkelstein, J.A., Marino, M.C., Ochs, H., Fuleihan, R., Scholl, P.R., Geha, R., Stiehm, E.R., and Conley, M.E. 2003. The X-linked hyper-IgM syndrome: Clinical and immunologic features of 79 patients. Medicine (Baltimore). 82(6):373–384. PMID: 14663287.
Neonatal chronic granulomatous disease with severe multisystemic inflammatory involvement
Roxane Labrossea, Guilhem Crosa, Helene Decaluwea, Colette Deslandresb, Francoise Le Deista, Isabel Fernandeza, Elie Haddada
aDivision of Clinical Immunology and Rheumatology, Pediatric Department, CHU Sainte-Justine, University of Montreal, Montreal, QC, Canada; bDivision of Gastroenterology, Pediatric Department, CHU Sainte-Justine, University of Montreal, Montreal, QC, Canada
Chronic granulomatous disease (CGD) is a primary disease of phagocytic function caused by defects in any subunit of the nicotinamide adenine dinucleotide phosphate oxidase. It is characterized by variable degrees of immunodeficiency and dysregulation of inflammatory responses, leading to granuloma formation and inflammatory disease.
We describe the case of a male infant diagnosed with CGD at 48 h of life because of positive family history. Antenatal ultrasounds revealed polyhydramnios, bilateral hydronephrosis, a hyperechogenic gut, and ascites. Within the first weeks of life, he developed a clinical picture compatible with gastric outlet obstruction confirmed with an abdominal ultrasound. Inflammatory markers were high with a CRP (C-reactive protein) at 89.4 mg/L, absolute neutrophil count at 63.58 × 109/L, and eosinophils reaching 15.74 × 109/L. After excluding any potential infectious etiology, prednisone was switched to IV methylprednisolone at 2 mg/kg because of lack of clinical response, and progressive improvement was noted over the following weeks. To this day, despite his major improvement, the patient remains dependent of total parenteral nutrition because of insufficient per os intake. A bone marrow transplantation is planned in the near future as a curative measure.
To our knowledge, this is the first case of neonatal CGD with such precocious and severe symptoms of the gut and bladder and evidence of neonatal involvement. The peripheral neutrophilia and eosinophilia are also quite atypical, especially in the absence of an infectious trigger.
Could it have been avoided? HSV encephalitis secondary to a TLR defect
Hani Hadi
Department of Allergy, Asthma, and Immunology, Children’s Mercy Hospitals and Clinics, Kansas City, MO, USA
Introduction: Toll-like-receptors (TLRs) play a critical role in the innate immune detection of molecular structures and patterns. Among the various TLRs, TLR3 recognizes dsRNA, which is an important intermediary to viruses such as Herpes Simplex (HSV) Type 1. Given the clinical rarity and possible under-recognition of TLR deficiencies, this diagnosis can be challenging. We illustrate the difficulty in diagnosing this condition by highlighting an unfortunate case of a boy who presented with HSV-1 encephalitis.
Case Description: A 12-month-old male presented to a tertiary care center acutely unwell and in status epilepticus. Other than fatigue earlier that day, no other preceding symptoms were noted by family.
His past medical history revealed the occurrence of a truncal vesicular rash at day 7 of life. At that time, Tzanck smear was positive to HSV. Blood PCR revealed 76 000 DNA copies/mL of HSV-1. A diagnosis of neonatal disseminated HSV was made and he was treated with IV acyclovir for 21 days during that admission. The Immunology service was consulted and they requested lymphocyte enumeration, naive/memory T cells, NK, and TLR function; however, these investigations were put on hold. He received oral antiviral prophylaxis for the next 5 months through the ID outpatient clinic, but eventually was lost to follow up.
With this re-admission, a full septic workup was initiated after stabilization. Althoguh his blood and urine cultures and PCR were normal, CSF was positive to HSV-1. Lymphocyte enumeration revealed T-cell lymphopenia (CD3 917mm3) but TREC was normal at 10 863 × 106 copies. TLR function revealed normal responses to TLR1-2, 4–8 stimulation, but a decreased cytokine response to TLR3 (Poly (I:C)) stimulation. With this information, gene sequencing was ordered for multiple genes, including MYD88, PIK3R1, STAT1, UNC93B1, and IRF7. While gene deletion and duplication for IRF7 and UNC93B1 genes were not identified, autosomal recessive heterozygous variants in both IRF7 and UNC93B1 were reported.
Unfortunately this patient suffers from severe loss of function, cognition, tone, mobility and communication. He remains fully NJ-tube dependent and has intractable epilepsy with frequent episodes of autonomic storming.
Discussion: This case highlights the need for ongoing and comprehensive evaluation in patients with unexplained “red flag” diagnoses. Recurrent and unusual viral infections should trigger an innate immune evaluation, and infection with HSV-1 should trigger a TLR functional assay. In addition, extensive discussion among clinicians involved in the care of medically complex patients is necessary to promote understanding and a rationale for further investigations. It is impossible to know if early diagnosis would have changed the eventual outcome in this patient; however, early diagnosis would have resulted in less distress to the patient, more informed decision making for the family, and less uncertainty to medical professionals during his acute representation.
Treatment of idiopathic pneumonia syndrome post-hematopoietic stem cell transplant with etanercept and rituximab in a STAT3-deficient hyper-IgE patient
Adam Byrne, Reza Alizadehfar, Christine Lejtenyi, Bruce Mazer, Christine McCusker
Division of Pediatric Allergy and Immunology, Department of Pediatrics, Montreal Children’s Hospital, McGill University Health Center, Montreal, QC, Canada
Background: Hyper-IgE syndrome (HIES) is a rare primary immunodeficiency with an unknown specific population prevalence. The autosomal dominant (AD) form of HIES, formerly known as Job syndrome, is most often caused by a loss of function mutation in the signal transduction and activation of transcription (STAT3) protein (Holland et al. 2007). The loss of STAT3 function alters cytokine response to foreign stimuli, leading to abnormal inflammatory patterns. Recurrent sinopulmonary infections from a young age lead to pneumatocele formation, providing a reservoir for the growth of opportunistic infections such as Pseudomonas aeruginosa and Aspergillus spp. (Freeman et al. 2007). It is thought that the abnormal inflammatory response to lower respiratory tract infections contributes to the pneumatocele formation, thereby leading to one of the more common causes of morbidity in AD-HIES patients. Alternatively, recurrent skin abscesses, usually due to Staphylococcus aureus, are described as “cold abscesses” due to the lack of associated inflammation (Eberting et al. 2004). The Staphylococcus aureus infections frequently exacerbate the severe eczema associated with AD-HIES patients. Mucocutaneous candidiasis is another example of an infectious manifestation indicating poor immune responses in AD-HIES patients. The elevated IgE concentrations that give the disease its name can range from 1000 IU/mL to 50 000 IU/mL (Joshi et al. 2009).
AD-HIES is also associated with a variety of extra-immune manifestations. AD-HIES presents with a characteristic facies, including a broad nose, deep-set eyes, a prominent forehead, and coarse facial features (Freeman and Holland 2009). Other musculoskeletal abnormalities include craniosynostosis, pathological fractures, osteoporosis, and retained primary teeth associated with palatal mucosal changes (Domingo et al. 2007; Heimall et al. 2010). Vascular abnormalities include coronary and intracranial aneurysms (Heimall et al. 2010). The mortality risk of AD-HIES patients is usually associated with secondary pulmonary infections (Freeman et al. 2007) or lymphomas (Leonard et al. 2004).
The main goal of treatment for AD-HIES is infection control (Yong et al. 2012). Surveillance for respiratory infections must be high, and treatment should be started quickly with any suspicion of an infection while considering bacterial, viral, or fungal etiologies. Bleach baths or prophylactic antibiotics may be required to control skin infections. Though not uniform, some AD-HIES patients may have a humoral dysfunction, indicated by abnormal immunoglobulin concentrations or vaccine responses (Sheerin and Buckley 1991), possibly dictating the need for intravenous immunoglobulin treatment.
Despite these measures, patients with AD-HIES are at high risk for long-term complications. Allogeneic hematopoietic stem cell transplant (HSCT) has been reported several times in the literature with varying degrees of success (Goussetis et al. 2010; Patel et al. 2015; Yanagimachi et al. 2016). One of the earliest case reports focused on a 46-year-old male with AD-HIES who received a peripheral stem cell transplant from his HLA-matched sister for treatment of resistant lymphoma (Nester et al. 1998). The patient responded well to therapy, but presented 6 months post-transplant with dyspnea and bilateral pulmonary infiltrates. A lung biopsy revealed acute interstitial pneumonitis with no infectious causes discovered. Despite treatment with antibiotics, azathioprine, and prednisone, the patient died; early interstitial pulmonary fibrosis and diffuse alveolar damage were noted on autopsy.
Idiopathic pneumonia syndrome (IPS) is a rare condition that occurs after HSCT and has a high mortality rate of up to 79% in one pediatric study (Sano et al. 2014). IPS has 3 defining criteria that include evidence of widespread alveolar injury; absence of active lower respiratory infection; and an absence of cardiac dysfunction, acute renal failure, or iatrogenic fluid overload as a cause for pulmonary dysfunction (Panoskaltsis-Mortari et al. 2011). Pediatric risk factors for development of IPS include prior HSCT and acute GVHD (graft-versus-host disease) of grade II–IV, whereas adult risk factors include older age at transplant, a diagnosis of acute leukemia or MDS, total body irradiation, and severe acute GVHD (Sano et al. 2014). Though there is no known mechanism, IPS is thought to result from a combination of factors, including pre-transplant conditioning, immunological cell-mediated injury, and inflammatory cytokines. Current standard of care includes broad spectrum antibiotics and high dose corticosteroids. Recently, however, clinical trials have focused on the use of etanercept, a tumor necrosis factor alpha (TNF-α) inhibitor to treat IPS (Yanik et al. 2008). TNF-α is a pro-inflammatory cytokine, resulting in neutrophilic attraction, increased cell-mediated cytotoxicity, and increased MHC (major histocompatibility complex) expression (Panoskaltsis-Mortari et al. 2011). Donor-derived TNF-α is the main regulator of the immunological environment in the lung during the first 2 weeks post-transplant. Treatment with a combination of etanercept twice weekly for 4 weeks in conjunction with systemic steroids and broad-spectrum antimicrobial treatment resulted in improved outcomes for patients with IPS post-HSCT (Yanik et al. 2008).
Patient History: The patient, a 2-year-old male at presentation, immigrated to Canada from Haiti in October of 2008. Prior to his immigration, he had reportedly been admitted to the hospital in Haiti for septic arthritis and multiple lower respiratory tract infections, including pneumonia requiring bilateral chest tubes. From October to December of 2008, he was evaluated in the pediatric emergency department multiple times for infected eczema and pneumonias before being admitted for treatment of a septic knee. During his admission, his history of frequent infections, craniosynostosis, and severe eczema prompted an immunology consult.
On initial evaluation, our patient had elevated eosinophils at 3.99 × 109/L. His IgG was 18.6 (6.29–17.01 g/L), IgA 0.96 (0.70–3.69 g/L), IgM 1.38 g/L (0.36–1.36 g/L), and IgE was 221 040 µg/L (0–240 µg/L). In 2008, the vaccine response was normal except several S. Pneumoniae serotypes, all of which summarily improved with further vaccination. His B-cell and T-cell enumerations were normal (CD3+ 2144, CD4+ 889, CD8+ 1046, CD19+ 2406, NK Cells 418). His T-cell simulation assays were mildly decreased in the context of an elevated baseline. On the basis of his dramatically elevated IgE, further investigations were completed, leading to a confirmed STAT3 mutation (1144 C>T; R382W) and resultant diagnosis of autosomal dominant hyper-IgE syndrome.
Our patient’s disease progressed over the years, despite multiple interventions. He continued to suffer from recurrent skin and respiratory infections, including an intensive care stay requiring intubation. In October of 2011, he started IV Ig therapy after diagnosis with a right humeral osteomyelitis with associated pathological fracture. Though his sinopulmonary infections improved, he developed multiple invasive fungal infections as well as bacterial skin and blood infections that were responsive to antimicrobial treatment, but they would return shortly after the treatments stopped. In the context of his unrelenting infections, the decision was made to pursue a hematopoietic stem cell transplant.
An unrelated, fully matched, CMV-negative donor was found and transplant was started in September of 2015. The patient’s reduced intensity pretransplant conditioning including Busulfan, Anti-Thymocyte Globulin, Fludarabine, and Thiotepa. His platelets engrafted 11 days post-transplant, whereas his neutrophils engrafted at 17 days. He received Cyclosporine and MMF (mycophenolate mofetil) as GVHD prophylaxis. His post-transplant course was complicated by hypertension, mucositis, a S. aureus/Malassezia furfur skin superinfection, a S. epidermis bacteremia, and a reactivation of CMV. The patient did well overall, and was discharged home in October 2015.
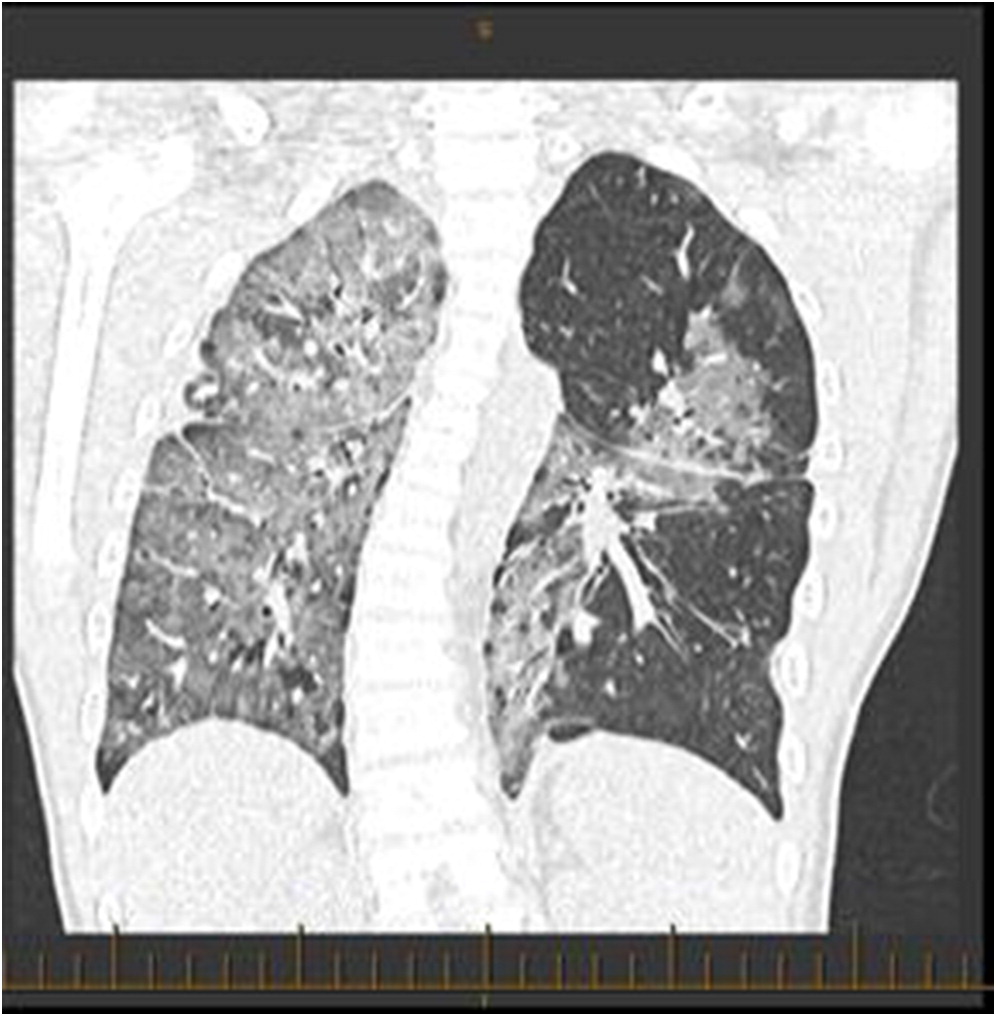
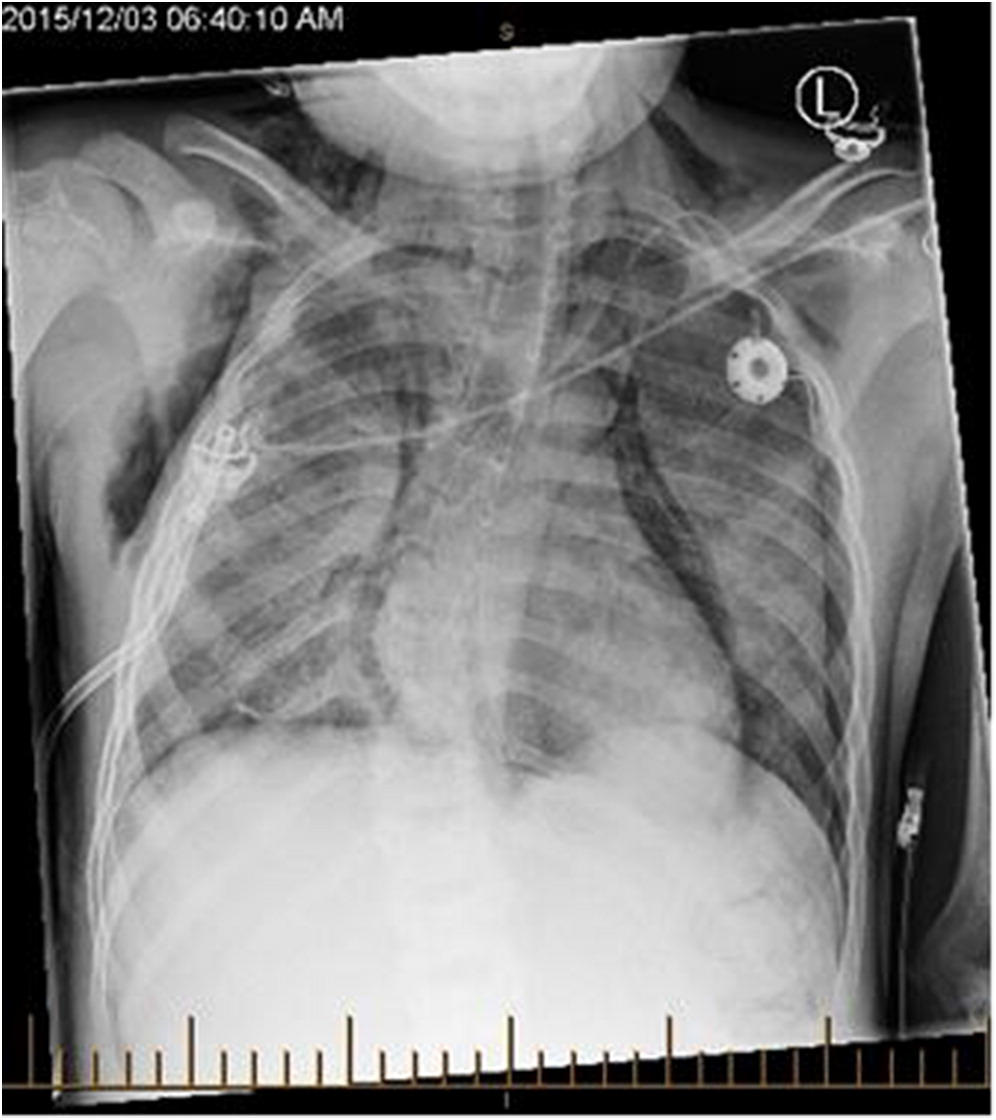
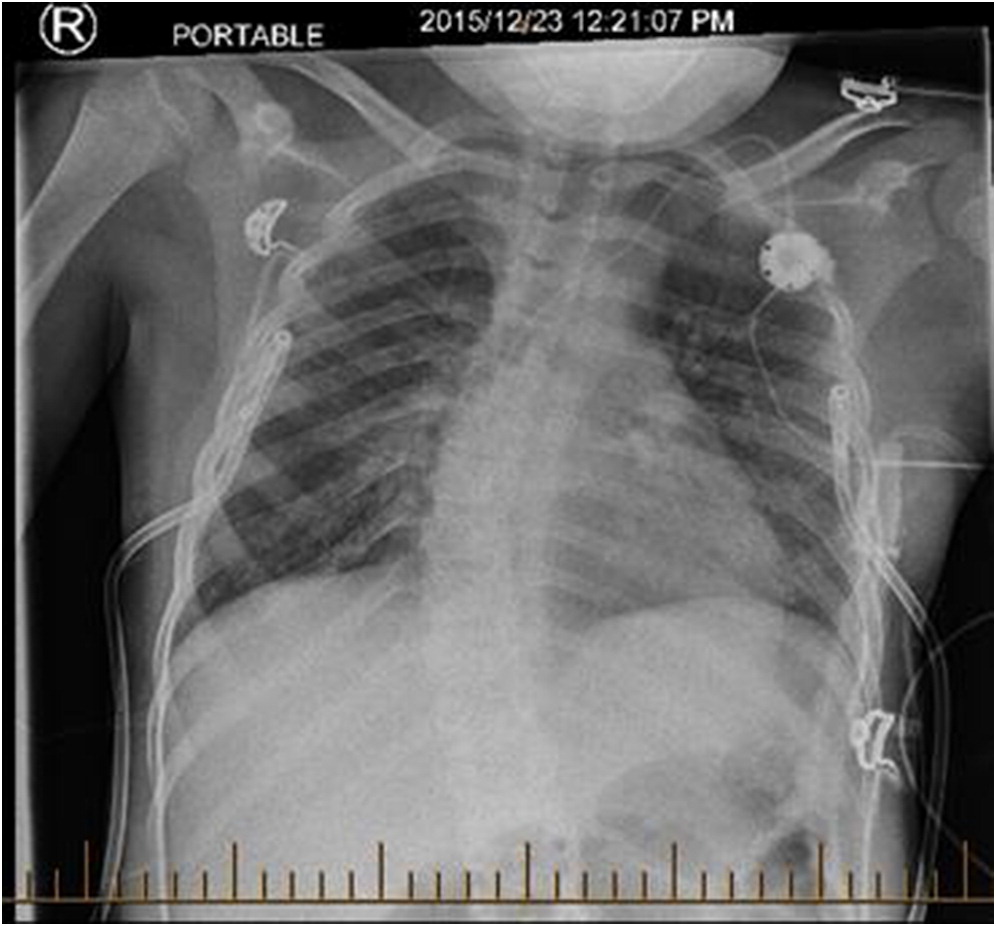
Several days after discharge, the patient returned with increased work of breathing and an elevated CMV PCR level. The patient clinically seemed to improve on gangciclovir treatment, and his improvement was associated with a decreased CMV PCR. By early November, however, his respiratory status had begun a gradual decline prompting transfer to the intensive care ward. A CT scan revealed “extensive groundglass opacities involving practically the entire right lung” with “similar but less pronounced groundglass opacities noted over the left perihilar region” (Figure 1). Multiple investigations were completed in the ICU, including a BAL and lung biopsy. The patient tested positive on BAL for BD glucan ((1,3) beta-D-glucan), and was started on pulse steroids and TMP-SMX (trimethoprim-sulfamethoxazole) for worries regarding a PCP infection. The lung biopsy demonstrated no PJP infection, however, and TMP-SMX was decreased to prophylactic dosing with a decrease in his steroid dosing. The lung biopsy did demonstrate “diffuse expansion of the interstitium by fibroblasts, early fibrosis and some mixed chronic active inflammatory infiltrate with prominent reactive pneumocytes and alveolar exudation”, all of which was suggestive of prominent interstitial pneumonitis. Infectious etiologies, including fungal cultures and HHV6 PCR, were reported as negative, and a consensus opinion was that our patient had IPS post-HSCT. Serum cytokines were analyzed, and the patient’s cytokines were generally normal, with a slightly elevated TNF-α and IL-6. Shortly after diagnosis, the patient then developed bilateral pneumothoraces and required BiPAP ventilation at high pressures (Figure 2).
Figure 1:
Figure 1: Patient chest CT. Demonstrates “extensive groundglass opacities involving practically the entire right lung. Similar but less pronounced groundglass opacities also noted over the left perihilar region”.
Figure 2:
Figure 2: Chest X-ray. This film taken the first day of pulse steroids/etanercept/rituximab treatment. Demonstrates a “right pneumothorax that has increased in size” and an “increase in pneumomediastinum” and an increase in the left and right subcutaneous emphysema. Diagnosed as a “severe pneumonitis that is unchanged from previous”.
With limited options and a deteriorating clinical picture, the patient was treated with pulse steroids and etanercept (0.4 mg/kg twice weekly for a total of 8 doses). Team discussions also led to a theory that there may be an element of our patient’s newly developing immune system activating a humoral autoimmune response against the pulmonary interstitium, so rituximab was also added to the treatment regimen at 375 mg/m2 weekly for 4 weeks. Palliative care was consulted. After 1 week of treatment with biologics, the patient had stabilized, and his BiPAP settings were gradually weaned. At 3 weeks after the start of biologics, the patient was transferred out of ICU on CPAP (continuous positive airway pressure) with improved chest imaging (Figure 3). After several months, the patient was discharged home on night-time CPAP, though his respiratory status remains quite fragile, and he is frequently readmitted with viral lung infections. A lung transplant was discussed with the family, and they refused.
Figure 3:
Figure 3: Chest X-ray. Image taken 20 days after initiation of pulse steroids/etanercept/rituximab.
REFERENCES
Domingo, D., Freeman, A.F., Davis, J., Puck, J.M., Tianxia, W., Holland, S.M., and Hart, T.C. 2007. Novel intraoral phenotypes in hyper immunoglobulin E syndrome. Oral Dis. 14:73–81. PMID: 18173452. doi: https://doi.org/10.1111/j.1601-0825.2007.01363.x.
Eberting, C., Davis, J., Puck, J.M., Holland, S.M., and Turner, M.L. 2004. Dermatitis and the newborn rash of hyper-IgE syndrome. Arch. Dermatol. 140:1119–1125. PMID: 15381553. doi: https://doi.org/10.1001/archderm.140.9.1119.
Freeman, A., Kleiner, D.E., Nadiminti, H., Davis, J., Quezado, M., Anderson, V., Puck, J.M., and Holland, S.M. 2007. Causes of death in hyper IgE syndrome. J. Allergy Clin. Immunol. 119:1234–1240. PMID: 17335882. doi: https://doi.org/10.1016/j.jaci.2006.12.666.
Goussetis, E., Peristeri, I., Kitra, V., Traeger-Synodinos, J., Theodosaki, M., Psarra, K., Kanariou, M., Tzortzatou-Stathopoulou, F., Petrakou, E., Fylaktou, I., Kanavakis, E., and Graphakos, S. 2010. Successful long-term immunologic reconstitution by allogeneic hematopoietic stem cell transplantation cures patients with autosomal dominant hyper-IgE syndrome. J. Allergy Clin. Immunol. 126:392–394. PMID: 20584545. doi: https://doi.org/10.1016/j.jaci.2010.05.005.
Holland, S.M., DeLeo, F.R., Elloumi, H.Z., Hsu, A.P., Uzel, G., Brodsky, N., Freeman, A.F., Demidowich, A., Davis, J., Turner, M.L., Anderson, V.L., Darnell, D.N., Welch, P.A., Kuhns, D.B., Frucht, D.M., Malech, H.L., Gallin, J.I., Kobayashi, S.D., Whitney, A.R., Voyich, J.M., Musser, J.M., Woellner, C., Schäffer, A.A., Puck, J.M., and Grimbacher, B. 2007. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 357(16):1608–1619. PMID: 17881745. doi: https://doi.org/10.1056/NEJMoa073687.
Joshi, A., Iyer, V.N., Boyce, T.G., Hagan, J.B., Park, M.A., and Abraham, R.S. 2009. Elevated serum immunoglobulin E (IgE): When to suspect hyper-IgE syndrome—A 10 year pediatric tertiary care center experience. Allergy Asthma Proc. 30(1):23–27. PMID: 19331717. doi: https://doi.org/10.2500/aap.2009.30.3193.
Leonard, G., Posadas, E., Herrmann, P.C., Anderson, V.L., Jaffe, E.S., Holland, S.M., and Wilson, W.H. 2004. Non-Hodgkin’s lymphoma in Job’s syndrome: A case report and literature review. Leuk. Lymphoma. 45:2521–2525. PMID: 15621772. doi: https://doi.org/10.1080/10428190400004463.
Nester, T., Wagnon, A.H., Reilly, W.F., Spitzer, G., Kjeldsberg, C.R., and Hill, H.R. 1998. Effects of allogeneic peripheral stem cell transplantation in a patient with job syndrome of hyperimmunoglobuliemia E and recurrent infections. Am. J. Med. 105:162–164. PMID: 9727824.
Panoskaltsis-Mortari, A., Griese, M., Madtes, D.K., Belperio, J.A., Haddad, I.Y., Folz, R.J., and Cooke, K.R. 2011. An official American Thoracic Society Research Statement: Noninfectious lung injury after hematopoietic stem cell transplantation: Idiopathic pneumonia syndrome. Am. J. Respir. Crit. Care Med. 183:1262–1279. PMID: 21531955. doi: https://doi.org/10.1164/rccm.2007-413ST.
Patel, N., Gallagher, J.L., Torgerson, T.R., and Gilman, A.L. 2015. Successful haploidentical donor hematopoietic stem cell transplant and restoration of STAT3 function in an adolescent with autosomal dominant hyper-IgE syndrome. J. Clin. Immunol. 35:479–485. PMID: 25962528. doi: https://doi.org/10.1007/s10875-015-0167-z.
Sano, H., Kobayashi, R., Iguchi, A., Suzuki, D., Kishimoto, K., Yasuda, K., and Kobayashi, K. 2014. Risk factor analysis of idiopathic pneumonia syndrome after allogeneic hematopoietic SCT in children. Bone Marrow Transplant. 49:38–41. PMID: 23955635. doi: https://doi.org/10.1038/bmt.2013.123.
Sheerin, K., and Buckley, R. 1991. Antibody responses to protein, polysaccharide, and phi X174 antigens in the hyperimmunoglobulin E (hyper-IgE) syndrome. J. Allergy Clin. Immunol. 87:803–811. PMID: 1826506.
Yanagimachi, M., Ohya, T., Yokosuka, T., Kajiwara, R., Tanaka, F., Goto, H., Takashima, T., Morio, T., and Yokota, S. 2016. The potential and limits of HSCT for the treatment of autosomal dominant hyper-IgE syndrome. J. Clin. Immunol. 36:511–516. PMID: 27091139. doi: https://doi.org/10.1007/s10875-016-0278-1.
Yanik, G.A., Ho, V.T., Levine, J.E., White, E.S., Braun, T., Antin, J.H., Whitfield, J., Custer, J., Jones, D., Ferrara, J.L., and Cooke, K.R. 2008. The impact of soluble tumor necrosis factor receptor etanercept on the treatment of idiopathic pneumonia syndrome after allogeneic hematopoietic stem cell transplantation. Blood. 112:3073–3081. PMID: 18664626. doi: https://doi.org/10.1182/blood-2008-03-143412.
Yong, P., Freeman, A.F., Engelhardt, K.R., Holland, S., Puck, J.M., and Grimbacher, B. 2012. An update on the hyper-IgE syndromes. Arthritis Res. Ther. 14:228–238. PMID: 23210525. doi: https://doi.org/10.1186/ar4069.
Activated PI3K-delta syndrome is a novel immunodeficiency caused by a gain-of-function mutation in PIK3CD
Hannah Roberts, Stuart Turvey
Division of Allergy and Clinical Immunology, Department of Pediatrics, University of British Columbia, BC Children’s Hospital, Vancouver, BC, Canada
Background: Activated PI3K-delta syndrome (APDS) or p110δ activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency (PASLI) is a rare autosomal dominant primary immunodeficiency (OMIM # 615513). It is caused by a heterozygous gain-of-function mutation in PIK3CD on chromosome 1p36.22. Since it was first described in 2013, only 30 patients with APDS from 17 unrelated families have been reported. We report a 13-year-old female who was found to have a de novo c.3061G>A PIK3CD gene mutation that is predicted to cause a glutamic acid-to-lysine substitution at codon 1021 (E1021K) of the p110δ protein to cause APDS (Jou et al. 2006; Angulo et al. 2013; Crank et al. 2014; Lucas et al. 2014).
Phosphoinositide 3-kinase (PI(3)K) classes phosphorylate the inositol ring of phosphatidylinositol lipids in membranes, and Class I PI(3)Ks have a role in immunity. P13K delta, a lipid kinase, consists of a catalytic p110δ subunit and a regulatory p85 subunit. The p110δ protein, encoded by PIK3CD, is involved in lymphocyte biology and adaptive immunity (Lucas et al. 2014). P13K delta is expressed primarily in cells of hematopoietic lineage and is involved in downstream signaling of B- and T-cell antigen receptors, cytokine receptors in T, B, and myeloid cells, and toll-like receptors (Angulo et al. 2013).
The clinical presentation and disease trajectory of APDS appears variable in the small population of patients reported to date, although a “typical” presentation can be summarized as lymphoproliferative common variable immunodeficiency (CVID) with increased risk of lymphoma. The majority of patients have been reported to have recurrent sinopulmonary infections in early childhood. Progressive airway damage and bronchiectasis has been reported. Other characteristics include lymphadenopathy, EBV and (or) CMV viremia, and deficiencies in T and B cells. The development of B-cell lymphomas has been described. Crank et al. (2014) reported 3 patients displaying classic hyper IgM phenotype in childhood and who as young adults developed malignant lymphoproliferative syndromes. Management strategies have included IgG substitution, antibiotics, immune modulation, and hematopoietic stem cell transplantation (Angulo et al. 2013; Crank et al. 2014; Lucas et al. 2014).
Methods: Clinical data were collected by retrospective chart review. The patient’s initial immunology assessment was in July 2007. In May 2016, our patient participated in the Clinical Assessment of the Utility of Sequencing and Evaluation as a Service exome sequencing study and results were diagnostic of APDS. As this is a recent diagnosis, the patient is due for reassessment in immunology clinic in September 2016. Further details gained directly from the patient at this clinic visit will be added to the complete presentation and manuscript.
Results:
Clinical features: The patient is a 13-year-old female who was born at term. She was initially assessed at 4 years of age by Immunology, prior to diagnosis of APDS. At the time, she had a recent CT scan (performed in Brazil) reported to show lung damage suggestive of bronchiectasis in the base or her right lung. This finding, in the setting of recurrent chest infections and wheezing episodes, initiated immunology assessment. She was well until 14 months of age. At that time, she began having multiple viral illnesses. From 14 months to 4 years of age, the primary complaint was persistent, bilateral rhinorrhea. Trials of nasal steroids and antihistamines made no improvement. The family reported long courses of antibiotics seemed to improve the rhinorrhea. Along with rhinorrhea, she had persistent wheezing and cough. Symptoms were worse at night and with physical activity. At age 4, she had been treated with beta-2 agonists and inhaled corticosteroids, which were reported to have no effect. She had an adenoidectomy that provided no significant benefit. On initial assessment, she had no history of acute otitis media, sinusitis, chronic oral candidiasis, or chronic diarrhea. Immunizations were up to date. There was no family history of immunodeficiency, immune dysregulation, or unexplained early deaths.
Since childhood, she has been followed by Respirology for upper respiratory tract infections, rhinorrhea, and bronchiectasis. She was reported to require a few courses of antibiotics over the years and had one admission for intravenous antibiotics. She was last assessed by Respirology in July 2016 and had no respiratory complaints, but she did have a recent ear infection requiring antibiotics. Pulmonary function testing at this time was normal. Vital capacity was 103% predicted (72% in 2010), and FEV1 was 114% predicted (79% in 2010). Medications consisted of fluticasone/salmeterol 125/25 mcg MDI 1 puff 3 times daily when unwell and methylphenidate 30 mg daily. Her growth and development have been normal. To date, she has had 1 episode of pneumonia, documented on chest X-ray, in June of 2007 and no history of meningitis or severe, unusual infections. On family history, her younger sister had an arachnoid cyst and underwent neurosurgery and shunt placement.
Immunological Features: When she was assessed at age 4, her CBC with differential was normal, except for a mildly low platelet count (WBC 6, neutrophils 2.94, lymphocytes 2.15, monocytes 0.66, eosinophils 0.21, basophils 0.02, hemoglobin 118, platelets 177). Serum immunoglobulin titers were normal (IgG 10.10 g/L, IgA 0.40 g/L, IgM 1.53 g/L, IgE 44). She had normal numbers of T cells, B cells, and NK cells. She had protective titers of antibodies for varicella zoster virus, diphtheria, and tetanus vaccines. Along with this, she had 2 negative sweat tests and negative epicutaneous testing to environmental allergens.
Conclusions: We report on a now 13-year-old female with a de novo c.3061G>A (p.E1021K) PIK3CD gene mutation, which has been reported in other patients with APDS (Angulo et al. 2013). The E1021K mutation in the catalytic subunit results in gain-of-function causing enhanced membrane association and kinase activity of p110δ. To our knowledge, there have only been approximately 30 other patients reported with this rare condition. There appears to be a diverse spectrum of clinical and immunological phenotypes associated with APDS. Interestingly, our patient had normal standard clinical immunological investigations at age 4 years. As progressive airway disease and malignant lymphoproliferative syndromes have been reported in patients with APDS, anticipatory guidance for these patients is critical. Precision therapy for APDS is under investigation using a selective PI3Kδ inhibitor, expanding future treatment options for this patient.
REFERENCES
Angulo, I., Vadas, O., Garçon, F., Banham-Hall, E., Plagnol, V., Leahy, T.R., Baxendale, H., Coulter, T., Curtis, J., Wu, C., Blake-Palmer, K., Perisic, O., Smyth, D., Maes, M., Fiddler, C., Juss, J., Cilliers, D., Markelj, G., Chandra, A., Farmer, G., Kielkowska, A., Clark, J., Kracker, S., Debré, M., Picard, C., Pellier, I., Jabado, N., Morris, J.A., Barcenas-Morales, G., Fischer, A., Stephens, L., Hawkins, P., Barrett, J.C., Abinun, M., Clatworthy, M., Durandy, A., Doffinger, R., Chilvers, E.R., Cant, A.J., Kumararatne, D., Okkenhaug, K., Williams, R.L., Condliffe, A., and Nejentsev, S. 2013. Phosphoinositide 3-kinase-delta gene mutation predisposes to respiratory infection and airway damage. Science. 342:866–871. PMID: 24136356. doi: https://doi.org/10.1126/science.1243292.
Crank, M.C., Grossman, J.K., Moir, S., Pittaluga, S., Buckner, C.M., Kardava, L., Agharahimi, A., Meuwissen, H., Stoddard, J., Niemela, J., Kuehn, H., and Rosenzweig, S.D. 2014. Mutations in PIK3CD can cause hyper IgM syndrome associated with increased cancer susceptibility. J. Clin. Immun. 34:272–276. PMID: 24610295. doi: https://doi.org/10.1007/s10875-014-0012-9.
Jou, S.-T., Chien, Y.H., Yang, Y.H., Wang, T.C., Shyur, S.D., Chou, C.C., Chang, M.L., Lin, D.T., Lin, K.H., and Chiang, B.L. 2006. Identification of variations in the human phosphoinositide 3-kinase p110-delta gene in children with primary B-cell immunodeficiency of unknown aetiology. Int. J. Immunogenet. 33:361–369. PMID: 16984281. doi: https://doi.org/10.1111/j.1744-313X.2006.00627.x.
Lucas, C.L., Kuehn, H.S., Zhao, F., Niemela, J.E., Deenick, E.K., Palendira, U., Avery, D.T., Moens, L., Cannons, J.L., Biancalana, M., Stoddard, J., Ouyang, W., Frucht, D.M., Rao, V.K., Atkinson, T.P., Agharahimi, A., Hussey, A.A., Folio, L.R., Olivier, K.N., Fleisher, T.A., Pittaluga, S., Holland, S.M., Cohen, J.I., Oliveira, J.B., Tangye, S.G., Schwartzberg, P.L., Lenardo, M.J., and Uzel, G. 2014. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110-delta result in T cell senescence and human immunodeficiency. Nat. Immunol. 15:88–97. PMID: 24165795. doi: https://doi.org/10.1038/ni.2771.
A novel mutation in LIG4 in an infant with severe combined immunodeficiency with unique thymic histological features
Willa Liaoa, Chaim M. Roifmana,b
aDivision of Immunology and Allergy, Department of Pediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada; bThe Canadian Centre for Primary Immunodeficiency, The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children, Toronto, ON, Canada
Introduction: DNA ligase IV (LIG4) has an essential role in T- and B-cell development. LIG4, in association with XRCC4 and Cernunnos/XLF, is involved in the final step of the nonhomologous end-joining (NHEJ) repair pathway involved in V(D)J recombination. This process, which also involves RAG1, RAG2, Ku70, Ku80, DNA-PKcs, Artemis, and TdT, contributes to the extensive diversity seen in the formation of T-cell receptors and immunoglobulins.
Mutations in LIG4 cause a rare autosomal recessive condition called LIG4 deficiency (OMIM #606593). Since it was first reported in 1999, there have been less than 30 cases of LIG4 deficiency published in the literature, with significant phenotypic variability among described patients. Cases have reported varying degrees of combined immunodeficiency, radiosensitivity, malignancy, developmental delay, growth failure, microcephaly and cytopenia. The most severe immunologic phenotype presents as a T-B-NK+ severe combined immunodeficiency (SCID).
Methods: We report a patient with a novel mutation in LIG4 who presented in infancy with phenotypic features of SCID.
Results: Our patient is the second child born to consanguineous parents of Pakistani descent. She was born at 40 weeks gestation following an unremarkable pregnancy and delivery. She presented at 4 months of age with an acute multilobar pneumonia, and she was also noted to have severe eczema, failure to thrive, and microcephaly. Parainfluenza virus type III, Pseudomonas aeruginosa, and Pneumocystis jirovecii were isolated by broncheoalveolar lavage. Pseudomonas aeruginosa was also positive on blood culture.
Her immune work up was significant for lymphopenia with near-absent CD19+ cells, and reduced CD4+ and CD8+ cells. Immunoglobulin levels were low. There was no response to mitogen stimulation to PHA, conA, PWM, STA, and SpA.
Based on these findings, a diagnosis of SCID was made at 5 months of age. HLA typing was performed but no matched sibling donor or suitable matched unrelated donor could be identified. As our patient’s clinical status continued to decline, we decided to proceed with peripheral blood stem cell transplantation from a 4/6 haploidentical donor (the patient’s mother). Unfortunately, the patient deteriorated, and she died from respiratory failure prior to transplantation at 8 months of age.
The patient’s autopsy revealed a significantly underdeveloped thymus that lacked clearly defined cortical and medullary regions as well as Hassall’s corpuscles, a finding which has previously not been reported. Furthermore, the histopathology was significant for extensive lymphocyte depletion within the thymus, lymph nodes, and spleen.
Through whole-exome sequencing, we identified a novel homozygous mutation c.1102G>T in LIG4 resulting in a D368Y amino acid substitution.
Conclusion: LIG4 deficiency is a rare primary immunodeficiency with a high degree of clinical variability. Our report contributes new data to the literature on LIG4 deficiency by reporting a novel mutation in LIG4 as well as unique histological findings encompassing an underdeveloped thymus in a patient who presented with a SCID phenotype.
Primary immunodeficiency’s grand masquerade: leukocyte adhesion deficiency type 1 presenting with pyoderma gangrenosum
Andrew O’Keefe, Alison Haynes, Lesley Turner, Paul Dancey
Division of Pediatrics, Faculty of Medicine, Memorial University, St. John’s, NL, Canada
Background: Leukocyte adhesion deficiency type-1 (LAD-1), first described in 1975, is a rare disorder estimated to affect 1/10 000 000 patients. Autosomal recessive mutations in the ITGB-2 gene result in defects in, or deficiency of, the adhesion molecule CD18. Severity of infection is thought to be related to the degree of CD18 expression. These patients are classically thought to present with delayed separation of the umbilicus, recurrent bacterial infections (often of the skin or mucosal surfaces), and leukocytosis.
Case Description: Our index patient presents as a 7-year-old female with necrotic, bullous skin lesions, requiring admission to hospital for pain control. During her admission a skin biopsy was consistent with pyoderma gangrenosum (PG). She was investigated for underlying rheumatologic and gastrointestinal disease, with no underlying etiology identified. Despite treatment with oral prednisone and cyclosporine A, she continued to have recurrence of these lesions. During a subsequent admission for pain control, she was assessed by genetics, who ordered a panel assessing for other diseases associated with PG. They identified a homozygous mutation in ITGB-2, consistent with a diagnosis of LAD-1 (c.2070 del p.Asp690Glufs*25). Soon after her diagnosis, her 13-year-old brother began to develop similar pustules on his thighs. He was later identified to have the same mutation in ITGB-2. Both patients have had improvement in their symptoms with antibiotic prophylaxis.
Discussion: Primary immunodeficiency disease can present in unusual and unexpected ways, and is not always associated with recurrent infection. There exist few case reports of LAD-1 presenting with PG, but this has been described previously. LAD-1 should be considered in cases of unexplained PG, particularly in children.
Multiple intestinal atresia with combined immune deficiency: a case report
Alexandra Langlois, Reza Alizadehfar, Bruce Mazer, Christine McCusker
Division of Pediatric Allergy and Immunology, Department of Pediatrics, Montreal Children’s Hospital, McGill University Health Center, Montreal, QC, Canada
Background: Familial cases of multiple intestinal atresia (MIA) have been described previously (Mishalany and Der Kaloustian 1971; Guttman et al. 1973). A subgroup of MIA patients also have signs of combined immunodeficiency and are termed multiple intestinal atresia with combined immune deficiency (MIACI) (Moreno et al. 1990). MIACI has been described in unrelated families of multiple ethnicities including Saudi, Slavic, Italian, Bosnian, and French-Canadian (Rothenberg et al. 1995; Chen et al. 2013). Mutations in the tetratricopeptide repeat (TPR) domain 7A gene (TTC7A) were identified using whole genome sequencing and is associated with MIACI (Chen et al. 2013; Samuels et al. 2013). MIACI is a rarely described multi-organ/multi-system disease (Fernandez et al. 2014). Herein we report the clinical presentation and the immunological phenotype of a patient with MIACI referred to the Montreal Children’s Hospital (MCH).
Case Presentation: This patient was born to French-Canadian parents and there was no history of consanguinity. Familial history was significant for a paternal grandfather who had a pulmonectomy at 9 years of age and who required IVIG, although further diagnosis was not available as he passed away at age 47 years from pneumonia. Our patient had 2 half-brothers aged 4 and 7 years whose clinical histories were positive for recurrent acute otitis media. The 4-year-old brother required 3 sets of tympanostomy tubes. There was no history of autoimmunity or neoplasia.
The patient was born late preterm at 34 +5 weeks of gestation in a regional center, 24 h after membrane rupture, by spontaneous vaginal delivery. Pregnancy was notable for polyhydramnios. Mother was GBS positive and received IV ampicillin. Initial Apgar scores were 9–9 and the cord pH was 7.31.
He developed apneas and vomiting with feeds in the first hours of life. An abdominal X-ray performed in the regional center showed distension of the stomach, absence of intestinal air, and diffuse calcifications in the abdomen.
The patient was therefore transferred to the MCH on day of life (DOL) #2 for management and evaluation by surgery. On DOL#3, an exploratory laparotomy confirmed a pyloric atresia type I, meconium cyst with meconium peritonitis, antenatal transverse colon perforation, and possible jejunal perforation. The patient underwent pyloroplasty and colonic and jejunal resection (2.1 cm from transverse colon and 2 cm from jejunum resected) with primary re-anastomosis. An ileostomy was performed to divert from the colon given the concern for possible Hirschsprung disease. This was reversed a month later.
His evolution was marked by severe secretory diarrhea with intermittently bloody stools. This led to major electrolytic and volemic imbalances despite exclusive parenteral intake as high as 250 mL/kg/day at times. His albumin levels were low and he needed regular supplementation with albumin. He displayed poor weight gain possibly secondary to his profuse diarrhea.
The infectious history was positive for a nosocomial Klebsiella pneumoniae urinary tract infection associated with a urinary catheter. All other cultures were negative.
On DOL#61 serum immunoglobulins were measured because of persistence of high spiking fevers. Serum IgM, IgG, and IgA were undetectable despite low normal levels of albumin. Laboratory abnormalities included intermittent severe normocytic anemia requiring multiple transfusions and intermittent leukocytosis up to 44.3 × 109/L. Neutrophils were chronically elevated with neutrophilia as high as 43.74 × 109/L and lymphocytes were intermittently low, the lowest value being 1.27 × 109/L. Platelet levels were variable.
He developed a rapidly progressing hepatosplenomegaly and displayed dysautonomic symptoms including hyperthermia, tachycardia, and variable blood pressure. Further investigations including a transesophageal echo showed subaortic stenosis and moderate mitral regurgitation. He developed hypothyroidism diagnosed at 4 months of age with high thyroid stimulating hormone 72.39 mIU/L (normal range 0.34–5.60 mIU/L) and low free thyroxine 7.2 pmol/L (normal range 12.0–55.0 pmol/L). Of note, newborn thyroid function screen was normal.
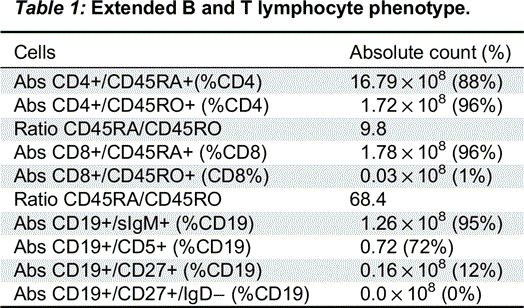
The immune evaluation showed that enumeration of B- and T-cell revealed decreased B cells (CD19 120 (5%), CD4 1778 (73%), CD8 200 (8%), CD4/CD8 Ratio 8.9, CD16_56 268 (11%)). An extended B and T lymphocyte phenotype showed high proportion of immature cells in both B and T cells with very little evidence of cell maturation (Table 1).
Table 1:
Table 1: Extended B and T lymphocyte phenotype.
A lymphocyte stimulation assay showed decreased proliferation to mitogens. Assessment of the V beta repertoire showed adequate variability.
The patient was persistently agammaglobulinemic. A dose of IVIG was given but IgG levels did not change and his clinical status remained precarious. Swallowed budesonide was given with mild clinical improvement in symptoms of esophagitis.
Genetic studies revealed compound heterozygous mutations in the TTC7A gene. One of the variant in the TTC7A gene was NM_001288951.1:c.1001+3_1001+6del(p.?). This variant causes a frameshift in exon 7 and is observed in less than 0.01% of patients (Broad ExAC dataset >60 000 individuals without severe childhood onset disease). This variant, reported in other cohorts, is considered pathogenic (Chen et al. 2013; Samuels et al. 2013). The second variant identified in the TTC7A gene of this patient is NM_001288951.1: c.518G>A(p.Gly173Asp) which, to our knowledge, has not previously been reported.
With the diagnosis of MIACI, based on clinical findings and genetic testing, a trial of regular SCIg was instituted. IgG levels increased to 11.44 g/L with SCIg (normal range 2.22–5.84) and dropped 2 days later to 0.86 and were undetectable 72 hours post initiation of SCIg despite doses as high as 200 mg/kg every other day. The palliative care service was consulted and given that the baby was still dependent of parenteral nutrition and the poor prognosis, the family elected to pursue comfort management only. The patient developed worsening respiratory distress and hypoxemia and passed away at 5 months of age, not long after diagnosis.
Conclusion: This patient was diagnosed with compound heterozygous mutations in the TTC7A genes. NM_001288951.1:c.1001+3_1001+6del(p.?), a known pathogenic mutation (Chen et al. 2013; Samuels et al. 2013) has been associated with disease MIA in both homozygous and heterozygous patients (Chen et al. 2013). This patient is the first known report of the compound heterozygous variants with a second mutation NM_001288951.1: c.518G>A(p.Gly173Asp). This patient presented with a severe clinical and laboratory picture of multiple intestinal atresia, severe agammaglobulinemia, decreased B cells, defects in lymphocyte maturation, and decreased proliferation to mitogens. The pathological diagnosis was MIACI. It is possible that the combination of mutations, this newly described variant plus the previously described “French-Canadian” mutation, led to MIACI.
REFERENCES
Chen, R., Giliani, S., Lanzi, G., Mias, G.I., Lonardi, S., Dobbs, K., Manis, J., Im, H., Gallagher, J.E., Phanstiel, D.H., Euskirchen, G., Lacroute, P., Bettinger, K., Moratto, D., Weinacht, K., Montin, D., Gallo, E., Mangili, G., Porta, F., Notarangelo, L.D., Pedretti, S., Al-Herz, W., Alfahdli, W., Comeau, A.M., Traister, R.S., Pai, S.-Y., Carella, G., Facchetti, F., Nadeau, K.C., Snyder, M., and Notarangelo, L.D. 2013. Whole exome sequencing identifies TTC7A mutations for combined immunodeficiency with intestinal atresias. J. Allergy Clin. Immunol. 132:656–664.e17. PMID: 23830146. doi: https://doi.org/10.1016/j.jaci.2013.06.013.
Fernandez, I., Patey, N., Marchand, V., Birlea, M., Maranda, B., Haddad, E., Decaluwe, H., and Le Deist, F. 2014. Multiple intestinal atresia with combined immune deficiency related to TTC7A defect is a multiorgan pathology: Study of a French-Canadian-based cohort. Medicine (Baltimore). 93:e327. PMID: 25546680. doi: https://doi.org/10.1097/MD.0000000000000327.
Guttman, F.M., Braun, P., Garance, P.H., Blanchard, H., Collin, P.P., Dallaire, L., Desjardins, J.G., and Perreault, G. 1973. Multiple atresias and a new syndrome of hereditary multiple atresias involving the gastrointestinal tract from stomach to rectum. J. Pediatr. Surg. 8:633–640. PMID: 4752999.
Mishalany, H.G., and Der Kaloustian, V.M. 1971. Familial multiple-level intestinal atresias: Report of two siblings. J. Pediatr. 79:124–125. PMID: 5091250.
Moreno, L.A., Gottrand, F., Turck, D., Manouvrier-Hanu, S., Mazingue, F., Morisot, C., Le Deist, F., Ricour, C., Nihoul-Fekete, C., and Debeugny, P. 1990. Severe combined immunodeficiency syndrome associated with autosomal recessive familial multiple gastrointestinal atresias: Study of a family. Am. J. Med. Genet. 37:143–146. PMID: 2240032. doi: https://doi.org/10.1002/ajmg.1320370133.
Rothenberg, M.E., White, F.V., Chilmonczyk, B., and Chatila, T. 1995. A syndrome involving immunodeficiency and multiple intestinal atresias. Immunodeficiency. 5:171–178. PMID: 7749436.
Samuels, M.E., Majewski, J., Alirezaie, N., Fernandez, I., Casals, F., Patey, N., Decaluwe, H., Gosselin, I., Haddad, E., Hodgkinson, A., Idaghdour, Y., Marchand, V., Michaud, J.L., Rodrigue, M.-A., Desjardins, S., Dubois, S., Le Deist, F., Awadalla, P., Raymond, V., and Maranda, B. 2013. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J. Med. Genet. 50:324–329. PMID: 23423984. doi: https://doi.org/10.1136/jmedgenet-2012-101483.
A novel variant in the RNU4ATAC-associated Roifman syndrome in 2 Belgian siblings: a case report
Sarah De Schryvera*, Delfien Bogaertb*, Frans De Baetsb, Sabine Van Daeleb, Petra Schelstraeteb, Reza Alizadehfara, Bart P. Leroyc, Elfride Debaered, Melissa Dullaerse†, Filomeen Haerynckb†
aDepartment of Pediatric Allergy and Immunology, Montreal Children’s Hospital, Montreal, QC, Canada; bDepartment of Pediatric Immunology and Pulmonology, Jeffrey Modell Diagnosis and Research Centre, Ghent University Hospital, Ghent, Belgium; cDepartment of Ophtalmology and Center for Medical Genetics Ghent, Ghent University Hospital, Ghent, Belgium; dCenter for Medical Genetics Ghent, Ghent University hospital, Ghent, Belgium; eClinical Immunology Research Lab, Department of Pulmonary Medicine, Ghent University Hospital, Ghent, Belgium; *,†These authors contributed equally to this abstract.
Background: Roifman syndrome is a rare autosomal recessive disorder characterized by humoral immunodeficiency, facial dysmorphism, growth retardation, cognitive delay, spondyloepiphyseal dysplasia, and retinal dystrophy (Roifman 1997, 1999).
Nine cases have been reported to date and the phenotype has been expanded with renal tubular dysfunction, hypogonadotrophic hypogonadism, and noncompaction of the myocardium (de Vries et al. 2006; Gray et al. 2011; Mandel et al. 2001).
Because most reported cases of Roifman syndrome are among males, an X-linked recessive inheritance had been suggested. Candidate gene studies using targeted sequencing were unsuccessful in identifying causal variants until recently.
In 2015, however, Merico et al. (2015) identified compound heterozygous variants that disrupt highly conserved positions of the RNU4ATAC gene in 2 previously described affected siblings. RNU4ATAC is a small nuclear RNA gene located on chromosome 2q14a and is a major component of the minor spliceosome that is essential for minor intron splicing.
RNU4ATAC variants had been reported to cause a phenotypically distinct severe congenital disorder, microcephalic osteodysplastic primordial dwarfism, type I (MOPD1). However, of the 6 cases described with Roifman syndrome, all had one variant overlapping MOPD1 mutations, whereas the other variant overlapped a highly conserved structural element not previously described.
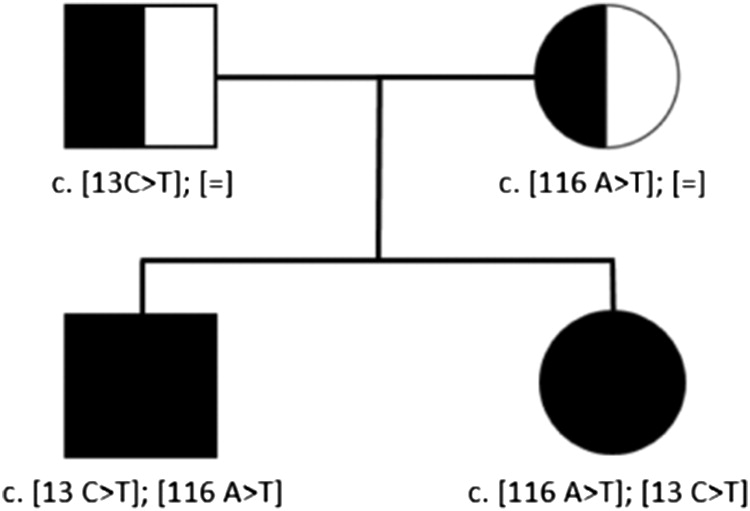
We have recently genetically identified an additional familial case of 2 Belgian siblings with a compound heterozygous mutation in RNU4ATAC with a previously described variant in the paternal allele (c.13C-T) and a novel variant in the maternal allele (c.116A-T).
Methods: Clinical data were collected by retrospective chart review. Informed consent was obtained. Genomic DNA was extracted from whole blood samples obtained from both patients and their parents. Whole exome sequencing and subsequent targeted Sanger sequencing was performed on the 2 affected siblings and their unaffected parents. The complete manuscript, currently in preparation, will describe the methods in detail.
Results: The patients, 2 siblings, a male and female currently 17 and 14 years old, respectively, are the only children of healthy, nonconsanguineous Belgian parents.
Clinical features: The boy initially presented at 18 months of age with a clinical picture of recurrent upper and lower respiratory tract infections and secondary bronchiectasis. The presence of hypogammaglobulinemia and poor response to polysaccharide and protein vaccines later on confirmed the diagnosis of CVID.
Additionally, at the age of 8 years, he gradually developed atypical clinical features of CVID with growth retardation and the following skeletal abnormalities: spondyloepiphyseal dysplasia lumbar hyperlordosis, “bullet-shaped” vertebrae, bilateral coax vara (femoral neck-shaft angle of 115°), and bilateral agenesis of the anterior cruciate ligaments and the 12th ribs. In addition, he developed mildly progressive retinal dystrophy with decreased rod function but normal cone function.
His sister presented similarly at 3 years of age with recurrent sino-pulmonary infections with subsequent bronchiectasis and generalized atopic dermatitis. In contrast, to date she has not developed any of the skeletal or retinal abnormalities. Both siblings developed the following mild dysmorphic features more apparent with age: short stature, long philtrum, thin upper lip, mildly tubular and upturned nose, mild microcephaly, and brachydactyly. None of them had signs of psychomotor delay.
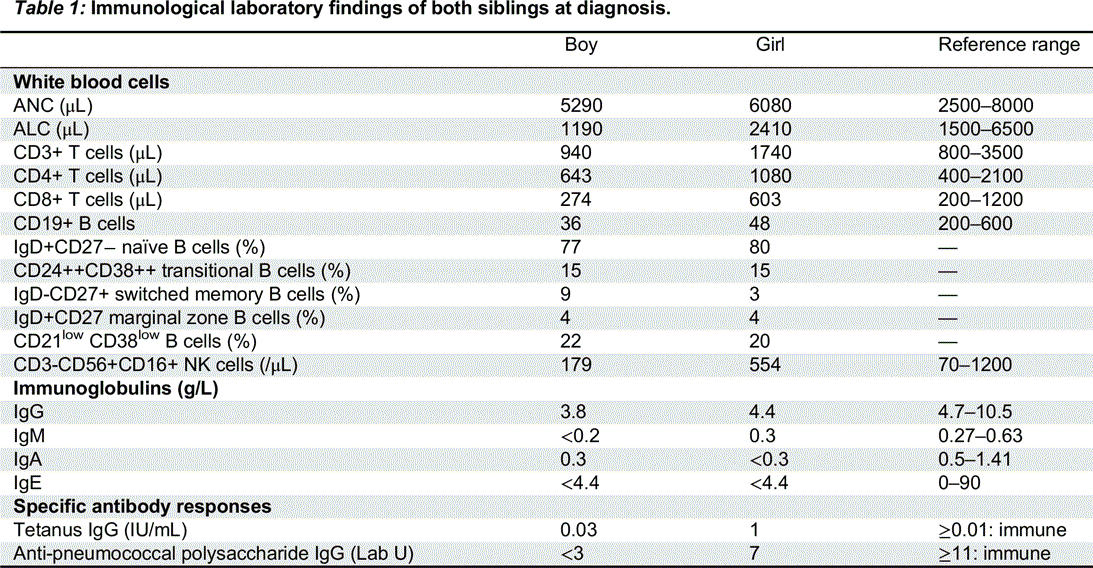
Immunological features: Immune work-up revealed a similar immunologic phenotype in both siblings, though it was more prominent in the male: decreased serum immunoglobulin levels and a lack of specific antibody response to polysaccharide vaccination. They presented with a severe B-cell lymphopenia with normal switched memory B cells and increased CD21low B cells. T-cell numbers and function were completely normal (Table 1).
Table 1:
Table 1: Immunological laboratory findings of both siblings at diagnosis.
Since the ages of 2.5 and 6.5 years in the male and female, respectively, they have been receiving monthly intravenous immunoglobulin (IVIg) and Azithromycin with a good clinical response. Based on the clinical history with accumulating clinical features and immunological assessment, the diagnosis of Roifman syndrome was suspected, especially in the male. However, the genetic diagnosis had not been confirmed until recently.
Whole-exome sequencing was performed on the 2 siblings and their parents. Since RNU4ATAC is a noncoding snRNA gene, possible variants would have been missed with WES. Subsequent Sanger Sequencing revealed compound heterozygous variants in RNU4ATAC in both siblings. Parents were each heterozygous for one of the variants. (Figure 1) The variant in the paternal allele (c.13C-T) had been previously described and associated with Roifman syndrome. In contrast, the variant in the maternal allele (c.116A-T) has not been previously reported.
Figure 1:
Figure 1: Familial pedigree showing the compound heterozygous mutations in the RNU4ATAC in the 2 affected siblings, parents were each heterozygous for one of the variants; [=] indicates no variant detected.
Conclusion: We present a familial case with a genetically confirmed diagnosis of Roifman syndrome in 2 siblings with a novel variant in the RNU4ATAC gene (c. 116A>T) that had not been reported previously. The girl presented with a milder clinical phenotype lacking the characteristic skeletal or retinal malformations, suggesting a possible broader clinical spectrum in patients with Roifman syndrome.
REFERENCES
De Vries PJ et al. 2006. The cognitive and behavioural phenotype of Roifman syndrome. J. Intellect. Disabil. Res. 50:690–696.
Gray et al. 2011. Is Roifman syndrome an X-linked ciliopathy with humoral immunodeficiency? Evidence from 2 new cases. Int. J. Immunogenetics. 38:501–505.
Mandel et al. 2001. Noncompaction of the myocardium associated with Roifman syndrome. Cardiol. Young. 11:240–243. PMID: 11293748.
Merico, D., Roifman, M., Braunschweig, U., Yuen, R.K., Alexandrova, R., Bates, A., Reid, B., Nalpathamkalam, T., Wang, Z., Thiruvahindrapuram, B., Gray, P., Kakakios, A., Peake, J., Hogarth, S., Manson, D., Buncic, R., Pereira, S.L., Herbrick, J.A., Blencowe, B.J., Roifman, C.M., and Scherer, S.W. 2015. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat. Commun. 6:8718. PMID: 26522830. doi: https://doi.org/10.1038/ncomms9718.
Roifman, CM. 1997. Immunological aspects of a novel immunodeficiency syndrome that includes antibody deficiency with normal immunoglobulins, spondyloepiphyseal dysplasia, growth and developmental delay, and retinal dystrophy. Canad. J Allergy Clin. Immunol. 2:94–98.
Roifman, CM. 1999. Antibody deficiency, growth retardation, spondyloepipheaseal dysplasia and retinal dystrophy: A novel syndrome. Clin Genet. 55:103–109. PMID: 10189087.
Mitogen proliferation assay reference ranges in patients with T-cell dysfunction
Mohammad Alsalamaha,b, Harjit Dadia,c, Linda Vonga,c, Chaim M. Roifmana,c
aDivision of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada; bPaediatrics Department, King Saud Bin Abdulaziz University for Health Science, University of Riyadh, Riyadh, Saudi Arabia; cThe Canadian Centre for Primary Immunodeficiency, The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children, Toronto, ON, Canada
Introduction: Mitogens are plant lectins that are potent stimuli for T-cell proliferation (Nowell 1960). For decades, assessment of lymphocyte proliferation has been a useful tool in the diagnosis of Severe Combined Immune Deficiency (SCID). Invariably, patients with SCID have low to absent responses to phytohemagglutinin (PHA) (Buckley et al. 1997). However, in patients with Combined Immunodeficiency (CID), the results of these tests have not been standardized and are open to a wide range of interpretations (Roifman et al. 2012). The primary goal of this study was to establish reference ranges for patients with T-cell defects, especially in those with CID. Identification of the gene defect associated with these T-cell disorders has allowed, for the first time, critical analysis of mitogen responses in the corresponding conditions.
Methods: Retrospective data were collected from 39 patients with SCID, 52 patients with CID, and 440 healthy controls. All participants had consented to the Primary Immunodeficiency Registry at The Canadian Centre for Primary Immunodeficiency (Protocol no. 1000005598). Genetic aberrations and lymphocyte proliferation responses to PHA, anti-CD3 antibody, and Staphylococcus Protein A (SPA) were compiled and used to calculate reference percentiles.
Results: For lymphocyte proliferation to the mitogen PHA, all 39 patients with SCID had a characteristically low stimulation index (SI) of between 0 and 5. In patients with CID, the SI ranged from 0.81 to 169. In contrast, the SI of healthy controls varied from 134 to 2220.
Anti-CD3 proliferation data from 4 patients with SCID showed SI <4. Thirteen patients with CID had a SI range from 0–50. Approximately 50% of healthy controls had a SI in the SCID/CID range of 0–50.
Results of lymphocyte proliferation to SPA show that majority of SCID patients (36) had a SI ranging from 0–10. For CID, 34 patients had a broad SI ranging from 0–400, thus overlapping widely with control samples.
Discussion: As previously described (Buckley et al. 1997; Roifman et al. 2012), when using PHA proliferation assay, all patients with typical SCID were found to have a profoundly low SI (range 0–5). Those with nontypical SCID (Omenn’s Syndrome or presence of maternal engraftment) also had a low SI (ranged from 0–30). The slightly higher SI in nontypical SCID is possibly due to a higher number of T cells (whether autologous or allogeneic) that may elicit detectable values. Almost all patients with CID (98%) had a SI of <200 and almost 85% had a SI <100. The SI of healthy controls demonstrated a wide range values, highlighting the difficulty of interpreting patient’s result based on relative comparison to control.
Conclusion: CID should be suspected in patients evaluated for underling PID with PHA mitogen assay SI of less than 200, regardless of the ratio to the healthy control. The results of this study delineate the statistically significant reference range of controls and patients with T-cell immunodeficiencies.
REFERENCES
Buckley, R.H., Schiff, R.I., Schiff, S.E., Markert, M.L., Williams, L.W., Harville, T.O., Roberts, J.L., and Puck, J.M. 1997. Human severe combined immunodeficiency: Genetic, phenotypic, and functional diversity in one hundred eight infants. J. Pediatr. 130(3):378–387. PMID: 9063412. doi: https://doi.org/10.1016/S0022-3476(97)70199-9.
Nowell, P.C. 1960. Phytohemagglutinin: An initiator of mitosis in cultures of normal human leukocytes. Cancer Res. 20:462–466. PMID: 14427849.
Roifman, C.M., Somech, R., Kavadas, F., Pires, L., Nahum, A., Dalal, I., and Grunebaum, E. 2012. Defining combined immunodeficiency. J. Allergy Clin. Immunol. 130(1):177–183. PMID: 22664165. doi: https://doi.org/10.1016/j.jaci.2012.04.029.
Hemizygous mutation in the Moesin gene is associated with profound lymphopenia and susceptibility to infections
Adi Ovadiaa, Yael Dinur Schejtera, Mohammad Alsalamaha, Stephen Feannya, Chaim M. Roifmana,b
aDivision of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada; bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children, Toronto, ON, Canada
The ERM (Ezrin, Ridixin, and Moesin) family of proteins function as cross-linkers between the plasma membrane and actin cytoskeletal structures. Moesin is localized to plasma membrane functional protrusions, such as filopodia or the T-cell receptor synapse, that are important for cell–cell recognition as well as cell mobility (Doi et al. 1999). Studies in lymphocytes suggest that Moesin helps to maintain the shape of circulating cells and contributes to cytoskeletal proliferation and relaxation (Parameswaran and Gupta 2013, Ivetic and Ridley 2004, Fehon et al. 2010, Pore and Gupta 2015).
We report here a 52-year-old male who had multiple invasive infections including meningitis at the age of 5 years, septic shock at 46 years, and multiple repeated episodes of cellulitis in the legs and face. At the age of 49 years he was diagnosed with Crohns disease. His immune evaluation revealed consistent lymphopenia with 470 CD3+ cells/µL, 280 CD4+ cells/µL, and 126 CD8+ cells/µL. CD19+ B cells as well as CD56+ NK cells were also reduced. Whole exome sequencing identified only one excellent candidate, a novel frameshift insertion in the Moesin gene (MSN) localized to exon 8 of the X chromosome. The change was not found in multiple large control databases and is predicted to act as an X-linked loss-of-function mutation. Very recently, other mutations in MSN were reported in patients with a similar phenotype to the patient described here.
Functional studies of lymphocyte mobility and adhesion will unravel the true impact of this mutation on Moesin function in the immune system.
REFERENCES
Doi, Y. et al. 1999. Normal development of mice and unimpaired cell adhesion/cell motility/actin-based cytoskeleton without compensatory up-regulation of ezrin or radixin in moesin gene knockout. J Biol Chem. 274(4):2315–2321. PMID: 9890997.
Parameswaran, N., Gupta, N. 2013. Re-defining ERM function in lymphocyte activation and migration. Immunol Rev. 256(1):63–79. PMID: 24117813. doi: https://doi.org/10.1111/imr.12104.
Fehon, R.G., McClatchey, A.I., Bretscher, A. 2010. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol 11:276–287. PMID: 20308985. doi: https://doi.org/10.1038/nrm2866.
Pore, D., Gupta, N. 2015. The ezrin-radixin-moesin family of proteins in the regulation of B-cell immune response. Crit Rev Immunol 35:15–31. PMID: 25746045.
2016. Abstracts from the Immunodeficiency Canada—4th SCID Symposium, Montreal, QC, 29 September 2016, and other Immunodeficiency Canada Conferences. LymphoSign Journal.
3(4): 179-201. https://doi.org/10.14785/lymphosign-2016-0010
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. A novel mutation in Roifman syndrome redefines the boundaries of the Sm protein-binding site
Note: This functionality works only for purchases done as a guest. If you already have an account, log in to access the content to which you are entitled.