Imaging in patients with chronic granulomatous disease
Abstract
Chronic granulomatous disease (CGD) is a primary immunodeficiency caused by defects in any of the subunits of nicotinamide adenine dinucleotide phosphate oxidase complex, required for proper phagocyte killing of bacteria and fungi. Most of the cases are X-linked, but autosomal recessive cases have also been identified. Patients suffer from recurrent, life-threatening infections and granulomatous inflammation of the skin, lymph nodes, lungs, liver, spleen, brain, and bones. CGD can be cured by hematopoietic stem cell transplantation. Imaging studies such as radiography, ultrasound, computed tomography, and magnetic resonance imaging play a key role in identifying the changes driven by both infection and dysregulated inflammation. These studies are critical for guiding management of this disorder. We present the most illustrative images from 7 patients with CGD.
Statement of novelty: Imaging studies are highly useful for diagnosis, treatment, and follow-up of patients with CGD. We present images from children with CGD that manifested their disease in different organs and tissues, illustrating the typical location of infections and dysregulated inflammation in these types of patients.
Introduction
Chronic granulomatous disease (CGD) is a primary immunodeficiency caused by defects in one of the subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex that results in the inability of phagocytes to generate superoxide, which is necessary for intracellular killing of both bacteria and fungi. These defects also predispose patients to granulomatous inflammation and autoimmunity (Battersby et al. 2013; Holland 2013; Roos and de Boer 2013.
Mutations in each of the 5 structural genes of the NADPH oxidase have been found to cause CGD. X-linked inherited mutations in gp91phox represent close to 65% of the cases, whereas autosomal recessive inheritance due to mutations in p47phox account for 25% and the remainder are divided between mutations in p67phox, p22phox, and the sole case of p40phox deficiency. There are no known autosomal dominant cases of CGD (Holland 2013).
The X-linked form is generally considered to be clinically more severe; patients have earlier presentation, more severe infections, and earlier death than the autosomal recessive forms (Holland 2013; Roos and de Boer 2013). However, CGD is a heterogeneous condition with some patients presenting early in infancy whilst others do not present until late childhood or even early adulthood, most likely a reflection of the degree of residual NADPH oxidase function (Battersby et al. 2013).
The most frequent sites of infection in CGD are the lung, skin, lymph nodes, and liver. The organisms causing the majority of these infections, at least in North America, are Staphylococcus aureus, Burkholderia cepacia complex, Serratia marcescens, Klebsiella species, Nocardia species, and Aspergillus species (Holland 2013). However, many other catalase positive organisms have been described in these patients.
Of them all, the most common site of infection are the lungs, with Aspergillus species being responsible for approximately 40% of the cases, although Staphylococcus aureus and Burkholderia cepacia are also important culprits (Kang et al. 2011). Liver abscesses with granulomatous inflammation occur in approximately 35% of cases and most are caused by Staphylococcus aureus. Up to 25% of patients suffer recurrence of these abscesses and they tend to develop multiple loculations, which makes percutaneous drainage difficult. The skin and lymph nodes are also involved in about 20% of patients, and the responsible infectious agents are usually Staphylococcus aureus, Klebsiella species, Serratia marcescens, Burkholderia cepacia, and fungi (Kang et al. 2011; Roos and de Boer 2013). Osteomyelitis is occasionally seen occurring either from hematogenous spread or contiguous invasion of bone.
Dysregulated granulomatous inflammation in CGD patients can be found in multiple tissues including the brain, lungs, liver, spleen, genitourinary tract, skin, and lymph nodes. Granulomas in CGD are typically noncaseating or nonnecrotizing and composed of multinucleated giant cells, and in most instances no pathogen is identified. Other complications associated with hyperinflammation experienced by some patients are conditions such as inflammatory bowel disease (IBD), lung fibrosis, and varied autoimmune conditions (Kang et al. 2011; Rieber et al. 2012; Holland 2013).
Granulomatous lesions in the lungs, liver, and spleen have been observed to wax and wane even without intervention. Moreover, clinical symptoms frequently do not alert to the true extent of tissue damage. It is therefore recommended to order imaging early in the disease (Towbin and Chaves 2010; Kang et al. 2011).
Diagnosis of CGD is initially made by measuring the NADPH oxidase activity in phagocytes either by the direct measurement of superoxide production, ferricytochrome c reduction, chemiluminescence, and nitroblue tetrazolium (NBT) reduction, or by dihydrorhodamine oxidation (DHR). Once the deficient NADPH oxidase activity is identified, confirmatory diagnosis is made by mutation analysis of the gene encoding for the subunits of this complex. The gene defect could be a predictor of the prognosis and, therefore, a useful tool in genetic counselling and prenatal diagnosis (Roos and de Boer 2013).
Prophylactic treatment with trimethoprim–sulfamethoxazole and an azole antifungal agent such as itraconazole is the most effective modality of treatment to reduce the frequency of potentially fatal bacterial and fungal infections. Nevertheless, despite appropriate prophylaxis, many patients still experience at least one severe bacterial or fungal infection every 3–4 years (Holland 2013; Roos and de Boer 2013).
Currently, the only cure for CGD is an allogeneic hematopoietic stem cell transplantation (HSCT) (Kang et al. 2011), and it is recommended to undergo this procedure as early as possible and prior to extension of end organ damage.
Gene therapy for CGD has not yet been proven to be a viable alternative to HSCT (Grez et al. 2011).
Cases
We present 7 illustrative pediatric cases of boys with CGD. Six of the patients had an X-linked form of CGD with confirmed mutations in CYBB, the gene encoding for gp91phox (Patients 1–6). The last patient (Patient 7) was shown to have a mutation in the NCF1 gene, encoding subunit p47phox of NADPH oxidase.
Patient 1
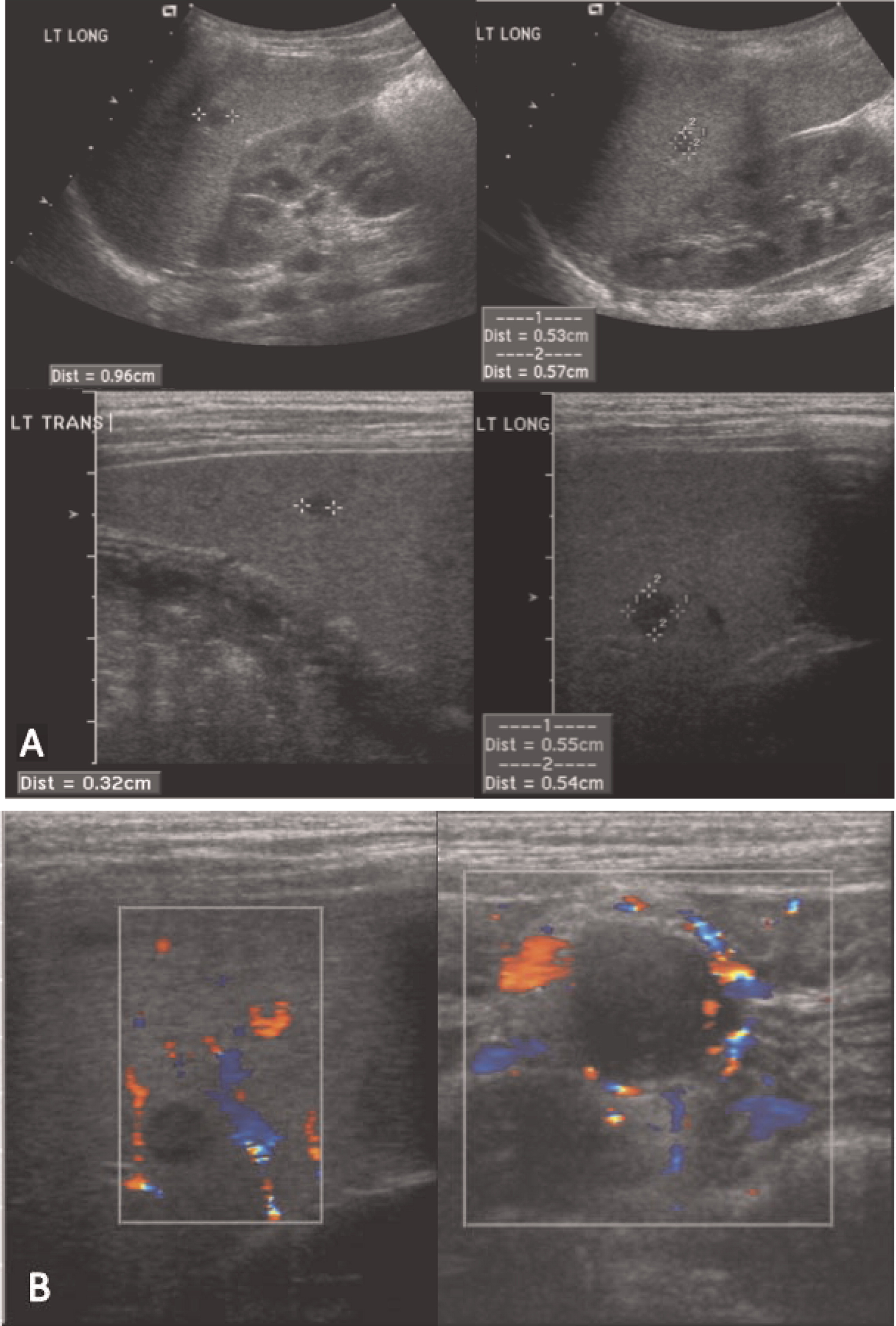
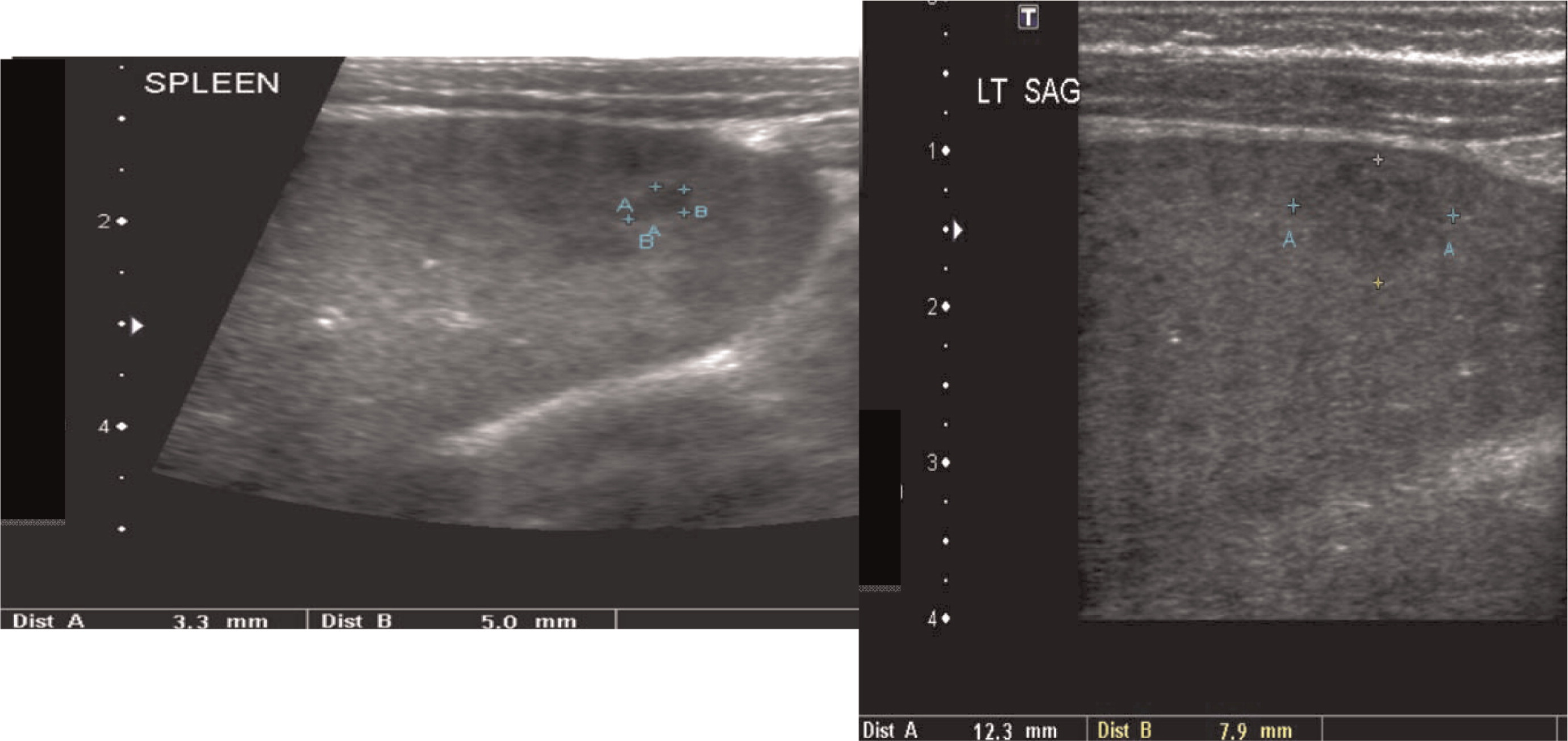
Patient 1 presented with a history of cervical lymphadenitis requiring excision at 1 year of age. He presented again at 3 years of age with an erythematous nodule with pustules on the medial aspect of his left arm with an accompanying enlarged lymph node in his left axilla (Figure 1). This lymph node was excised and found to grow Aspergillus in culture. This finding led to the suspicion and then confirmation of X-linked CGD. A computed tomography (CT) scan of his chest demonstrated mediastinal and hilar lymphadenopathy (Figure 2) as well as extensive nodular lung disease (Figure 3). He was treated with amphotericin and later with voriconazole and itraconazole. A follow-up CT scan after 4 months of antifungal treatment showed complete resolution of the axillary lesion and a significant decrease in the number of lung lesions with stable mediastinal and hilar lymphadenopathy. Six months after antifungal treatment was started, abdominal ultrasound (US, Figure 4) showed multiple splenic lesions and splenomegaly. He was treated again with amphotericin and caspofungin. These lesions waxed and waned over the following months. A laparoscopic biopsy was then performed, which was negative for fungi or bacteria but showed nonnecrotizing granulomas on microscopic examination. He was started on weekly interferon gamma therapy. He received a matched sibling donor HSCT and tolerated the procedure well. He was fully engrafted and all lesions completely resolved.
Figure 1:
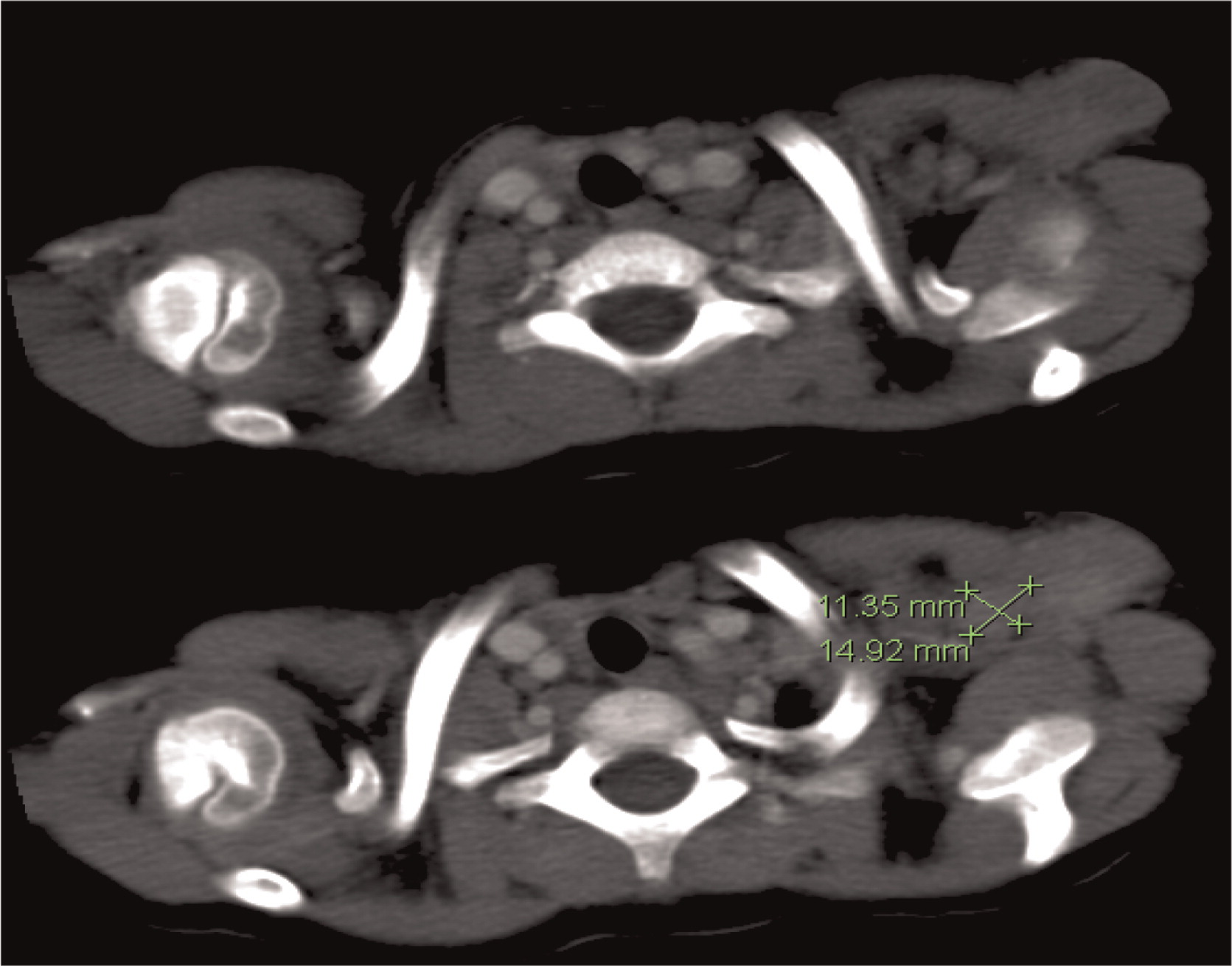
Figure 2:
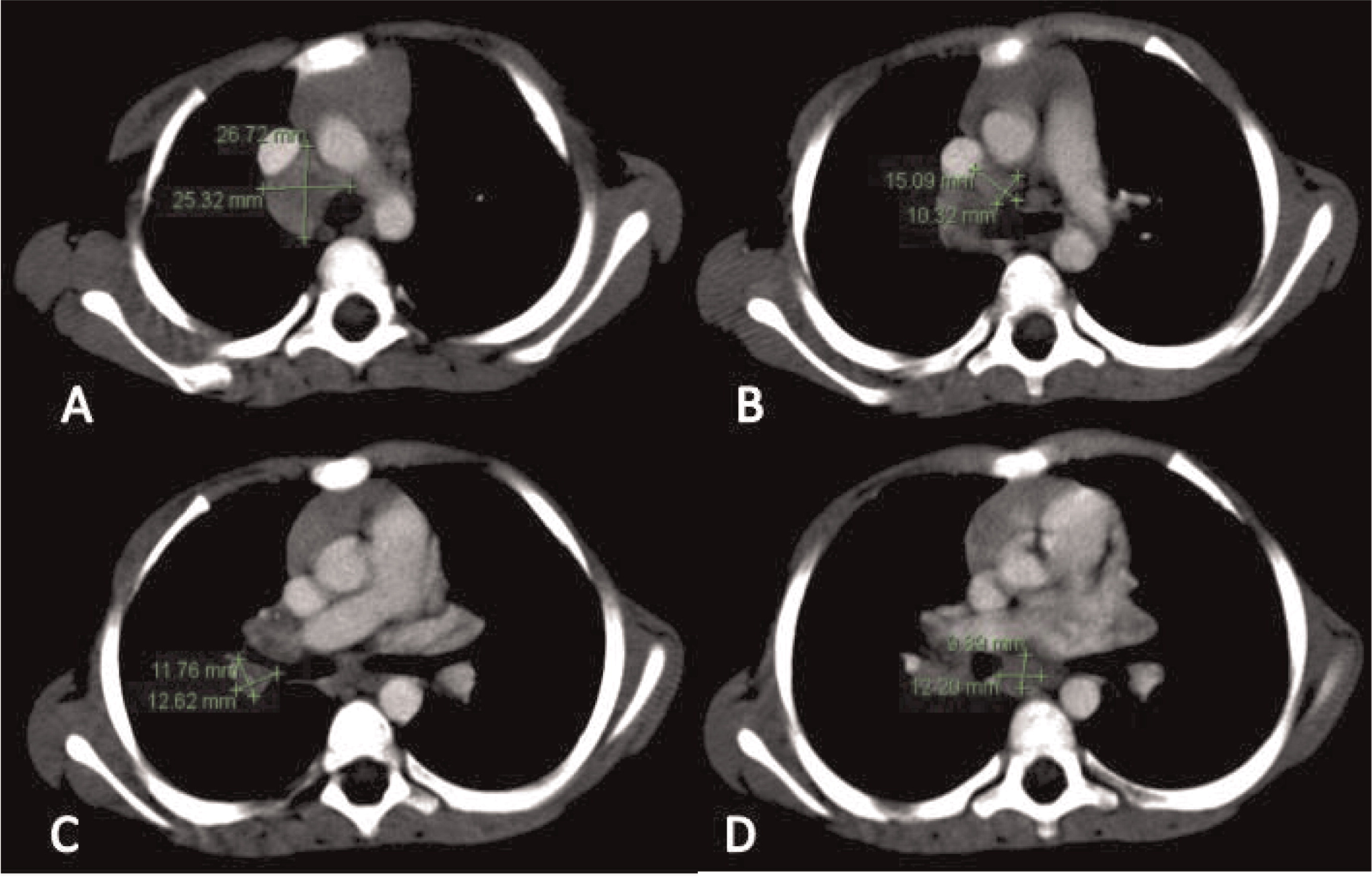
Figure 3:
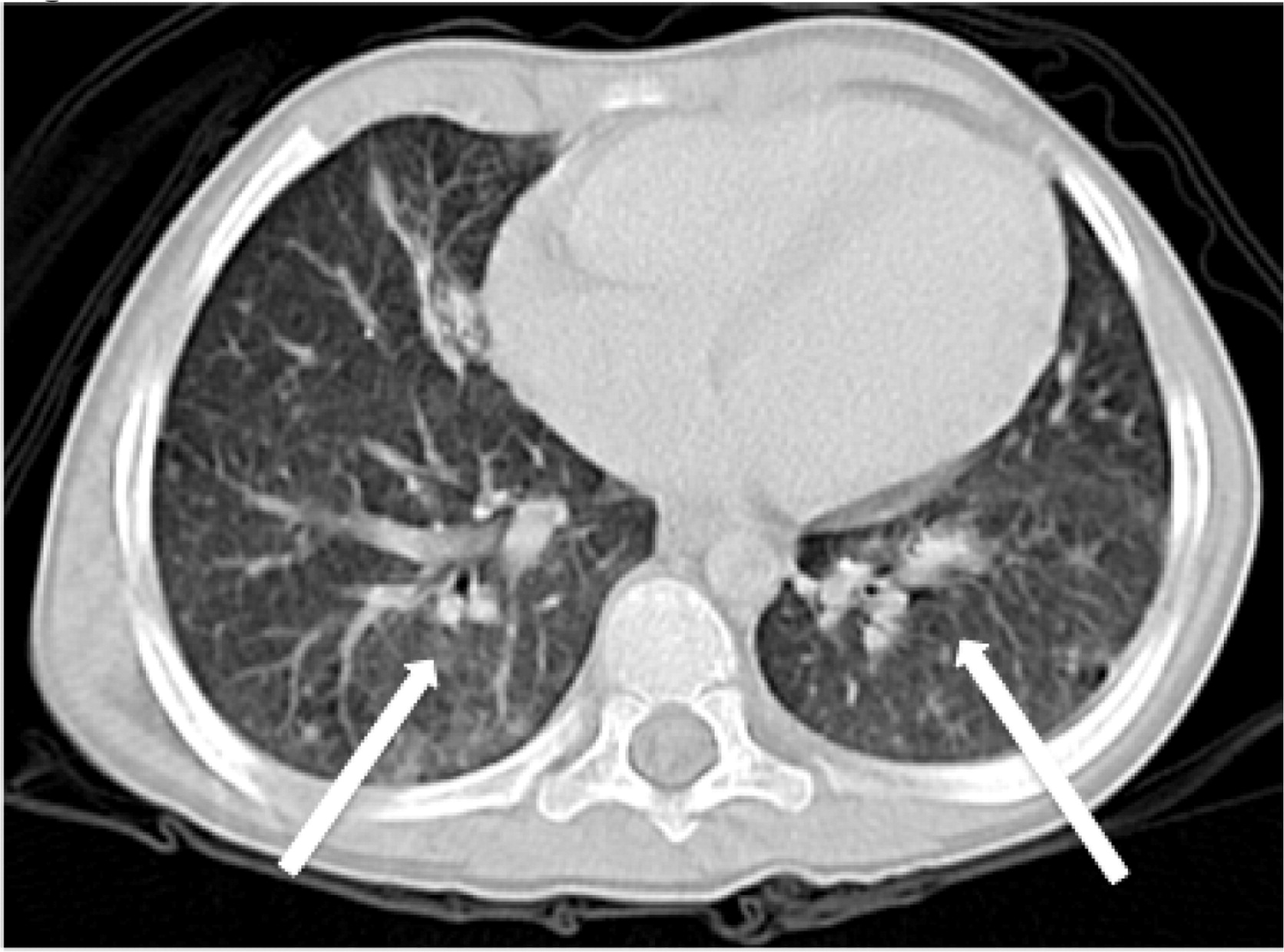
Figure 4:
Patient 2
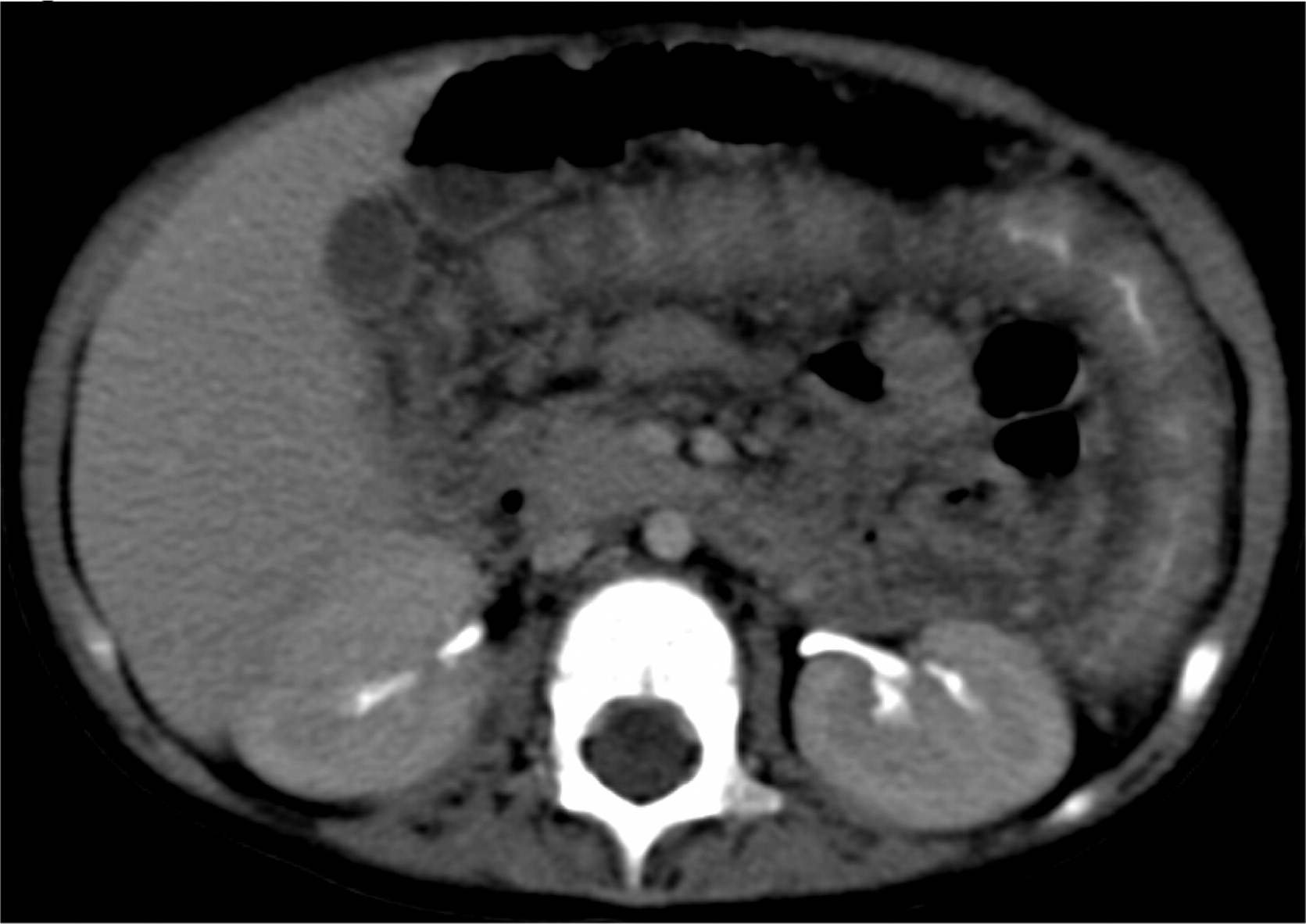
Patient 2 presented at 18 months of age with a 1-month history of diarrhea and fever. An abdominal US and CT scan were performed and showed 2 lesions in his liver (Figures 5 and 6). He was treated with cloxacillin and ciprofloxacin as well as prophylactic itraconazole. He had liver involvement and therefore underwent a biopsy that showed nonnecrotizing granulomatous inflammation (Figure 7). He continued to have persistent fevers and was eventually found to have a Serratia marcescens in a urine culture. US and CT of his abdomen showed bowel wall thickening (Figure 8). An upper and lower endoscopy showed severe colitis, more significant in the proximal than the distal bowel. He was given mesalamine as well as interferon gamma 3 times a week. A CT of his chest showed scattered small lesions in the posterior area of both lungs. Once the infections were controlled, he underwent a successful matched sibling donor HSCT.
Figure 5:
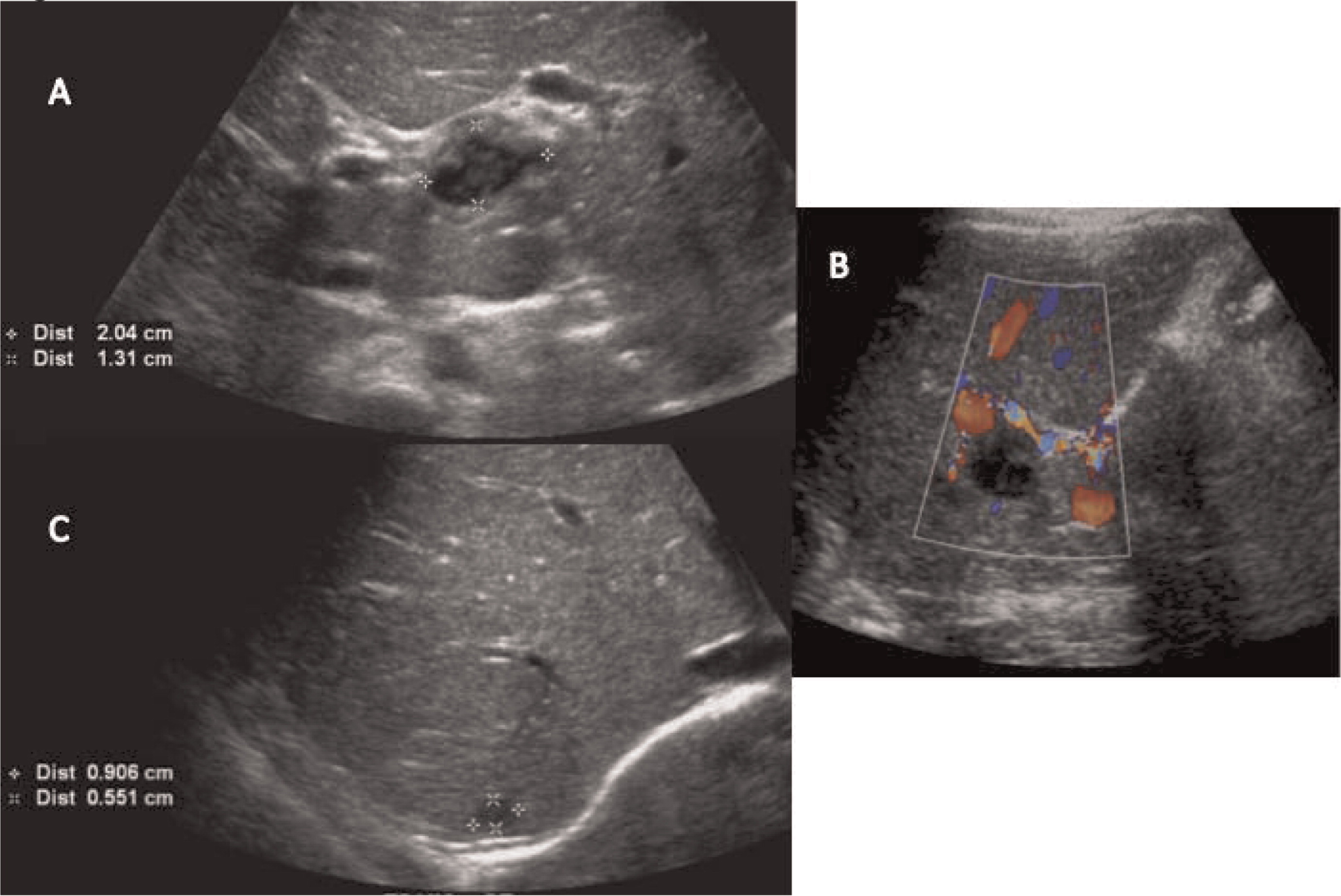
Figure 6:
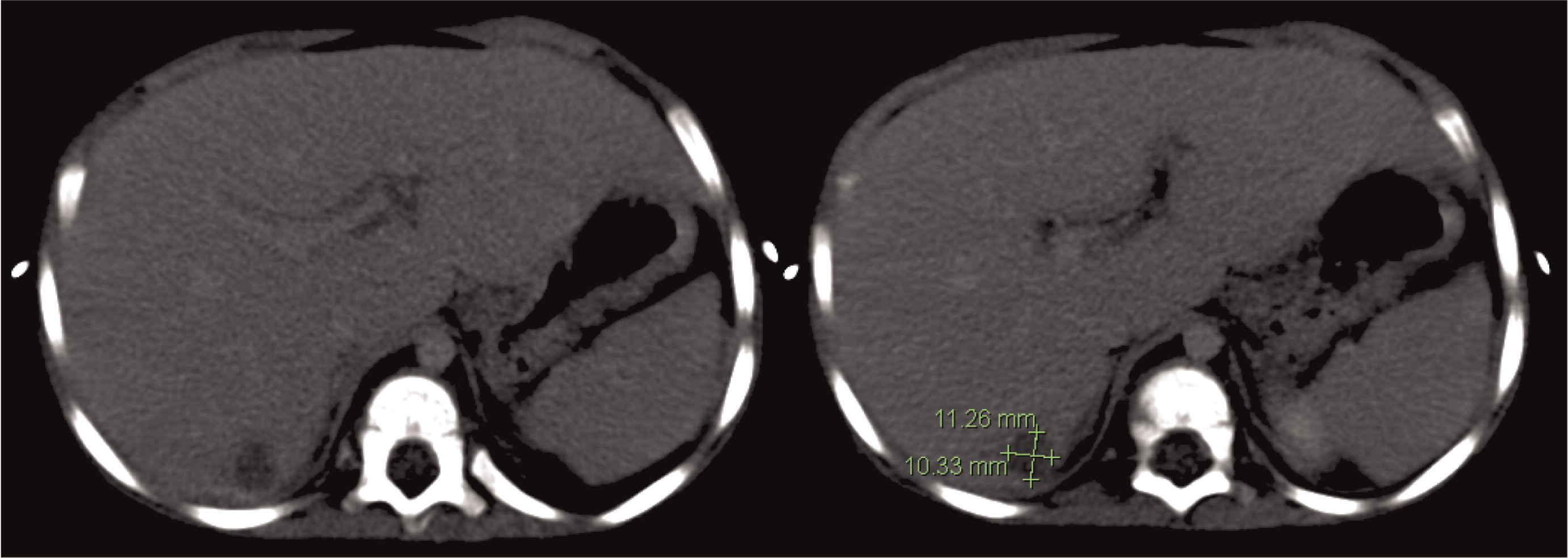
Figure 7:
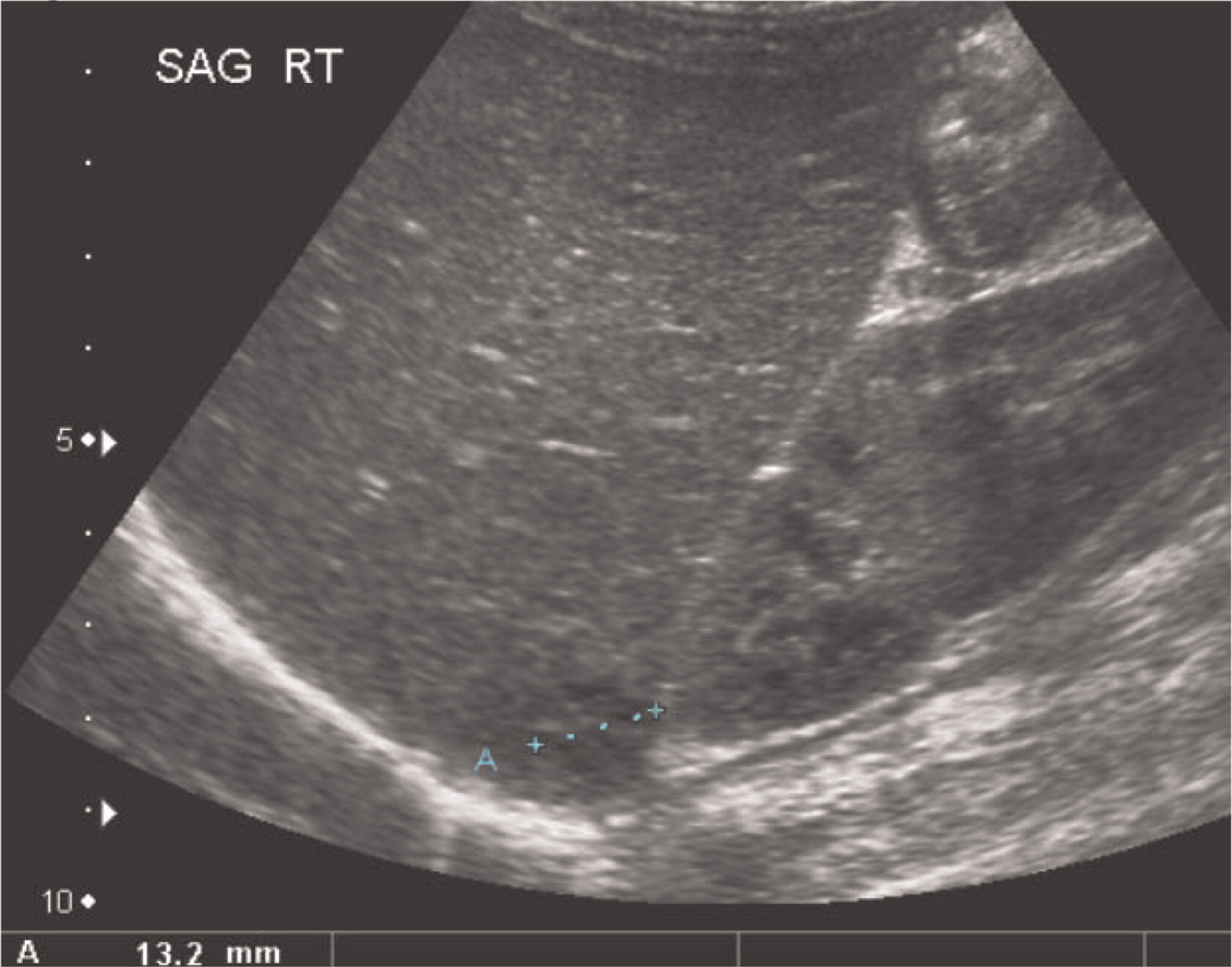
Figure 8:
Patient 3
Patient 3 presented initially at 2 months of age with left-sided cervical lymphadenopathy requiring excision and debridement. Cultures grew Staphylococcus aureus and microscopically the tissue showed granulomatous inflammation. He was treated but presented again at 4.5 months of age with a right-sided purulent postauricular lymphadenopathy (Figures 9 and 10) and persistent fevers. Cultures again grew Staphylococcus aureus and the microscopic description was again consistent with granulomatous inflammation. His infection was treated and the diagnosis of X-linked CGD was made. Given the persistence of fever, further imaging studies were performed. A CT of his chest showed multiple small pulmonary nodules (Figure 11) and an abdominal US revealed splenomegaly with focal splenic hypoechoic lesions (Figure 12). Once his lymphadenitis and fever resolved, he was discharged on trimethoprim–sulfamethoxazole and itraconazole prophylaxis, and he did well on this treatment. He underwent a matched sibling donor HSCT at 9 months of age with an uneventful course. Engraftment and immune reconstitution are robust with complete resolution of symptoms.
Figure 9:
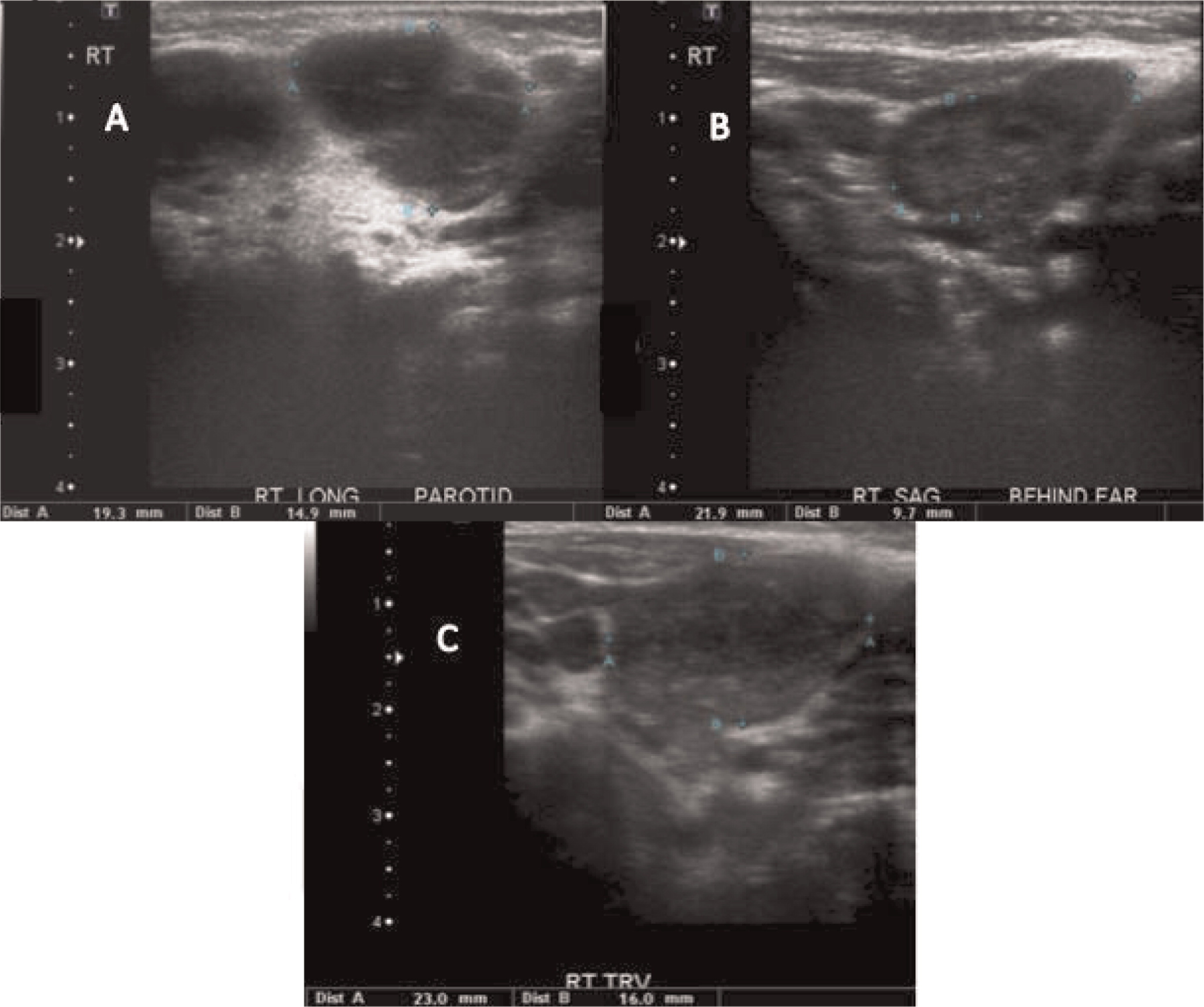
Figure 10:
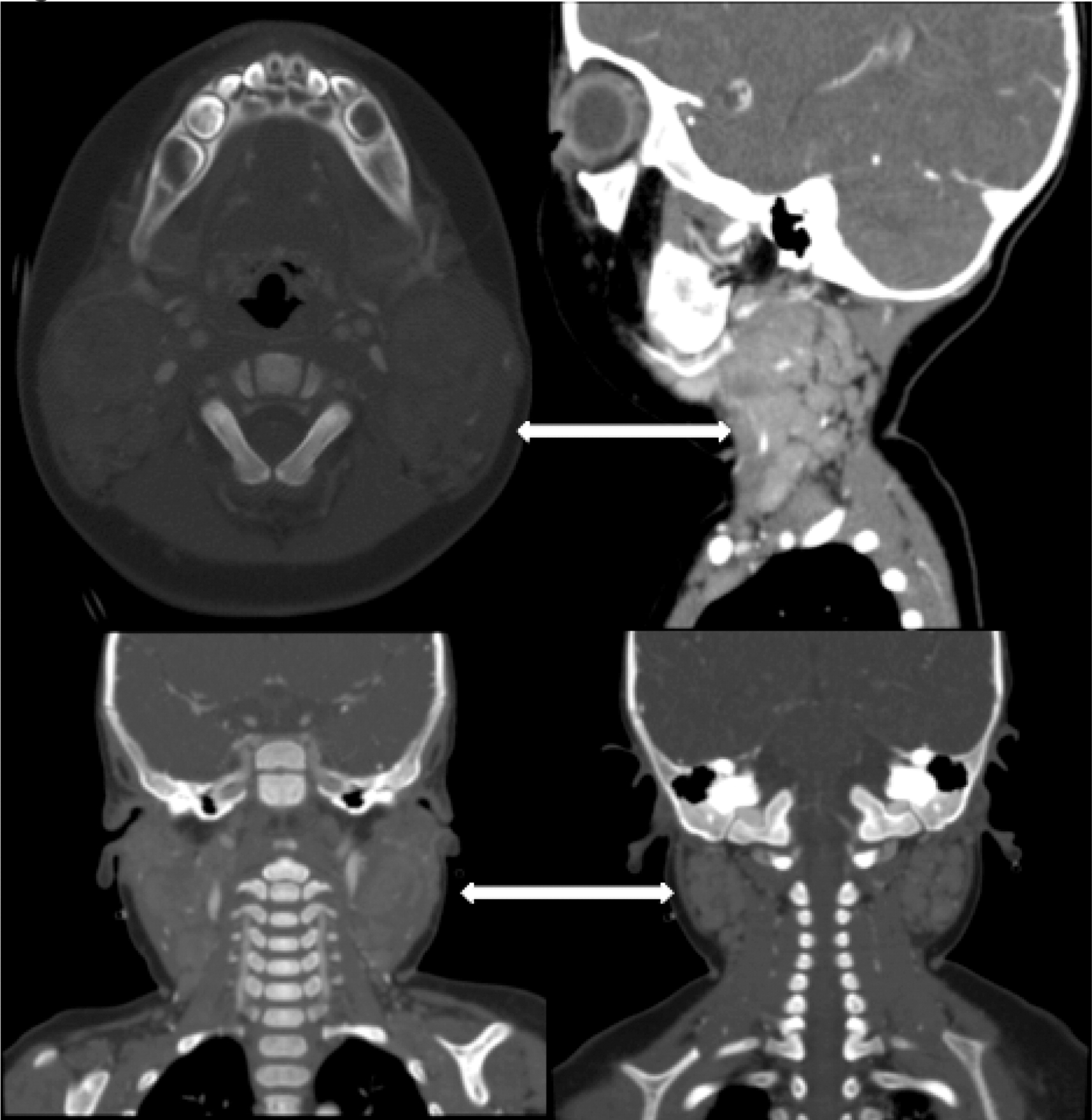
Figure 11:
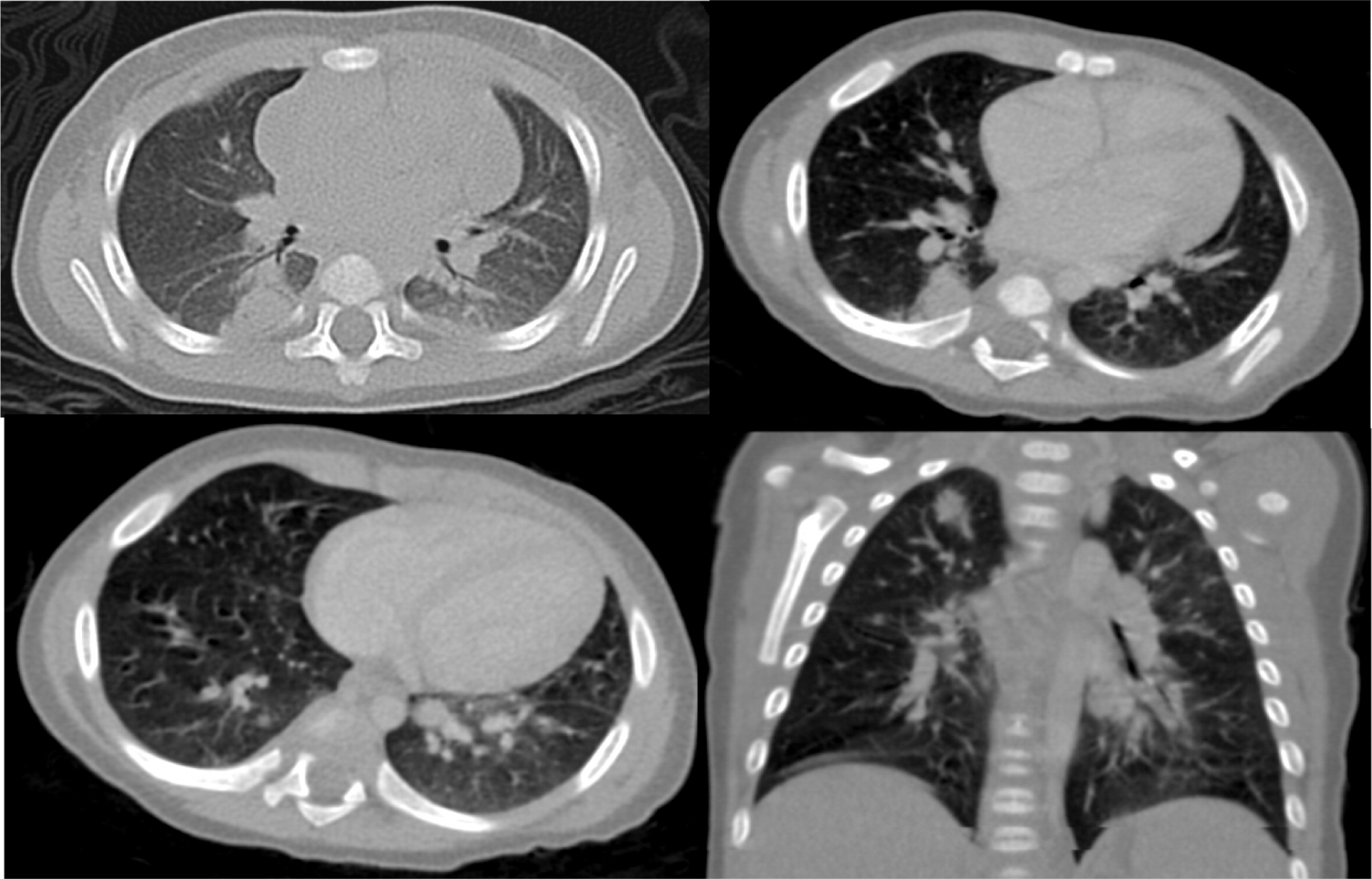
Figure 12:
Patient 4
Patient 4 presented at 5 months of age with persistent fevers, cervical lymphadenitis/abscesses (Figure 13), and liver abscesses (Figures 14 and 15). He had drainage of one of the cervical lymph nodes and a liver lesion, both growing Staphylococcus aureus and both showing granulomatous inflammation. Despite proper antibiotic coverage he had persistence of fevers. A CT scan of his chest showed lesions in his lungs (Figure 16). He was confirmed to have X-linked CGD. He underwent a successful matched sibling donor HSCT at the age of 8 months but unfortunately died of respiratory and cardiac arrest 5 months post-transplant.
Figure 13:
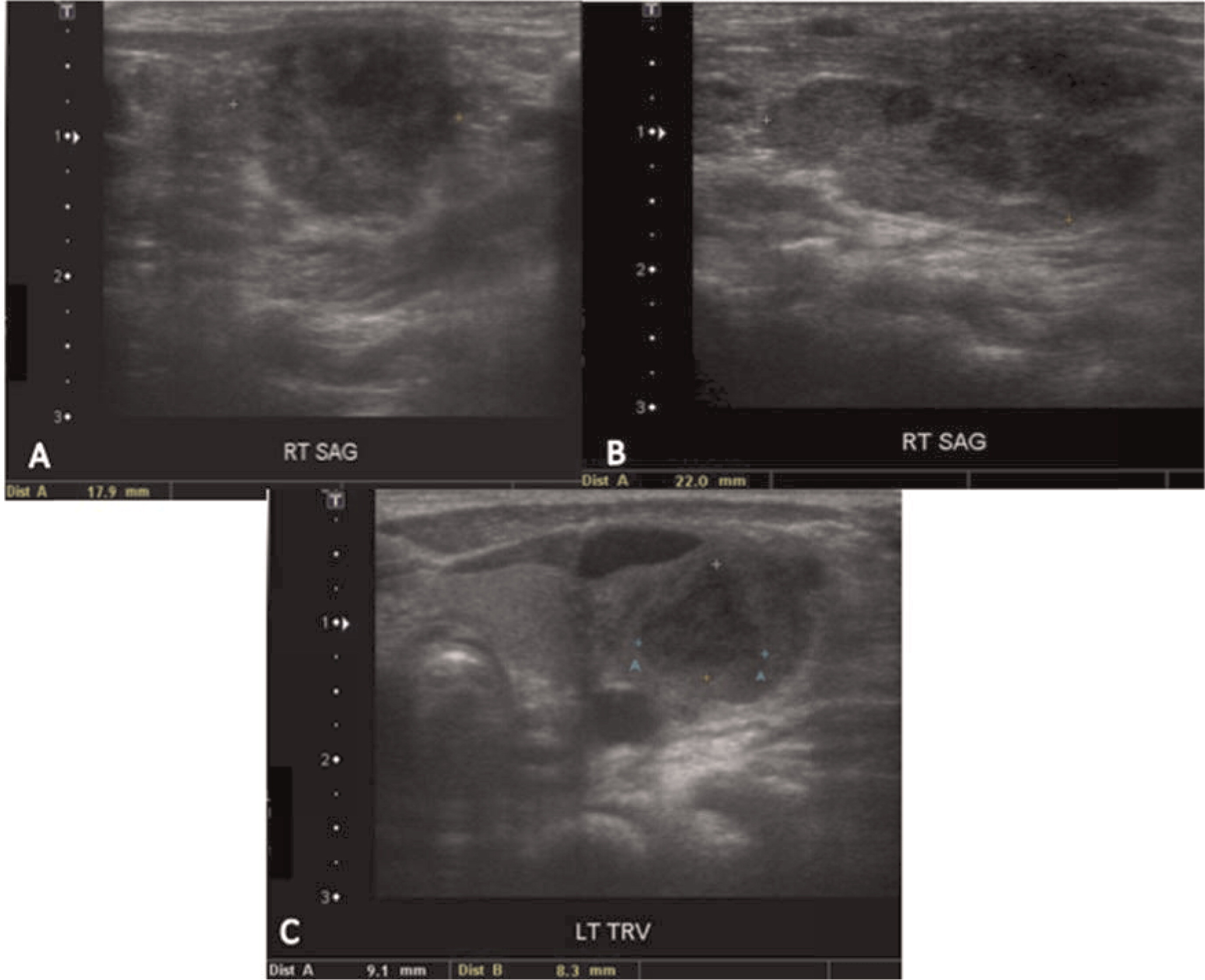
Figure 14:
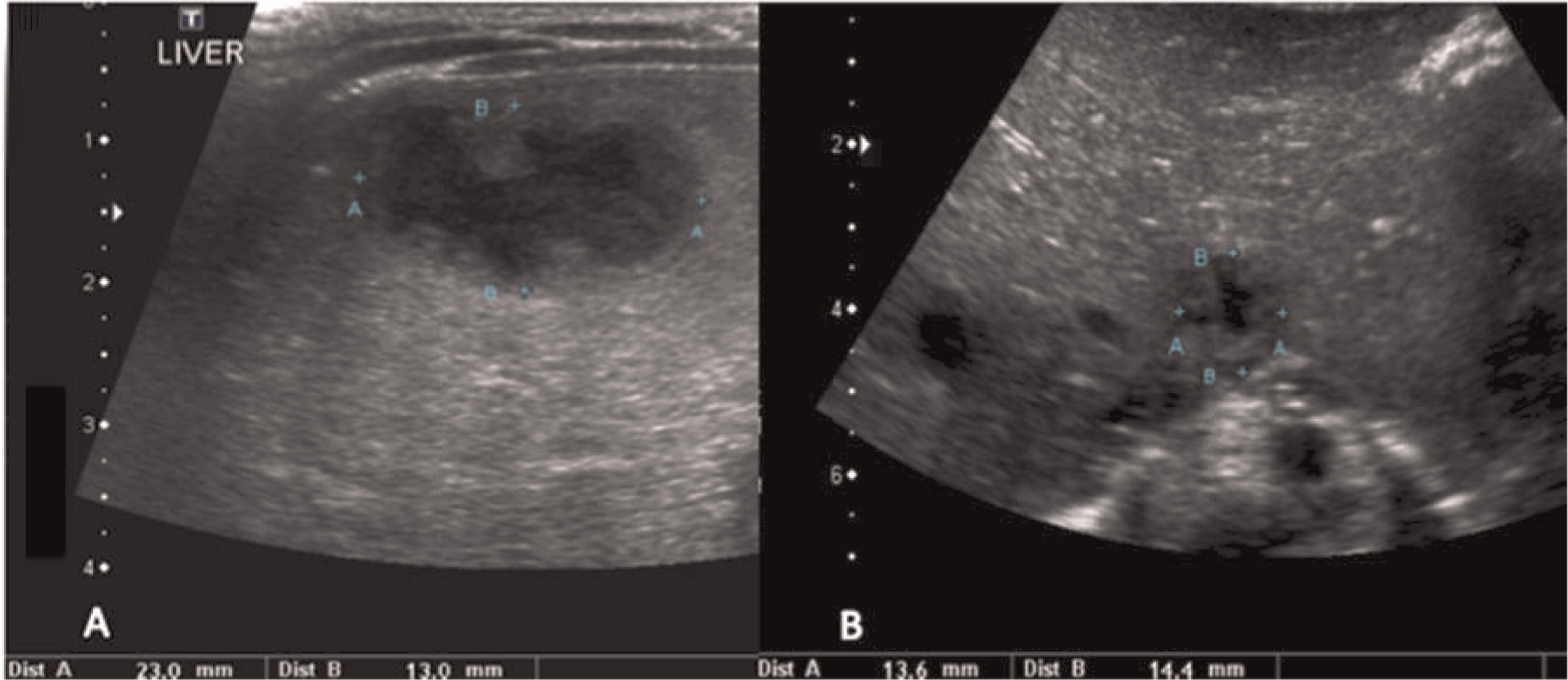
Figure 15:
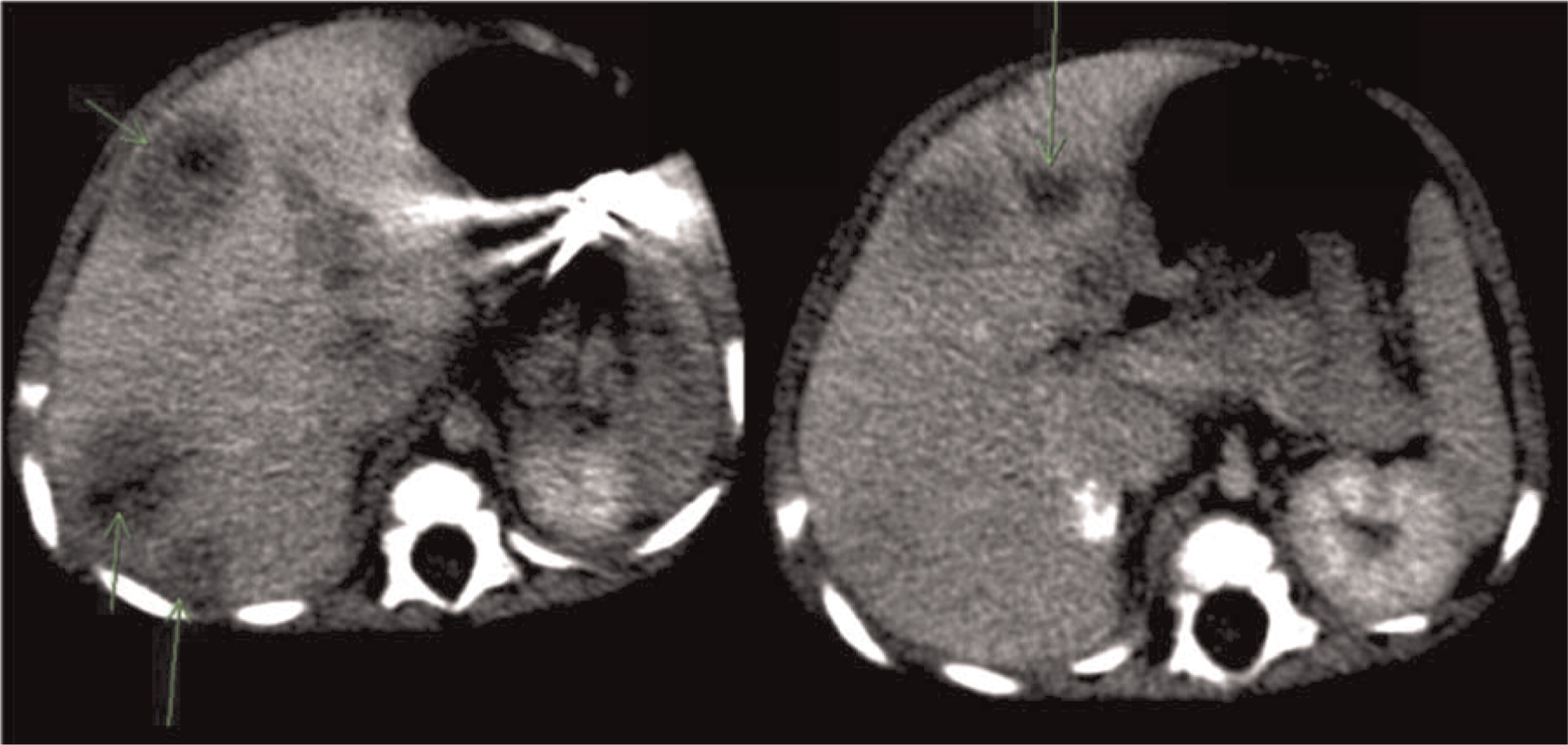
Figure 16:
Patient 5
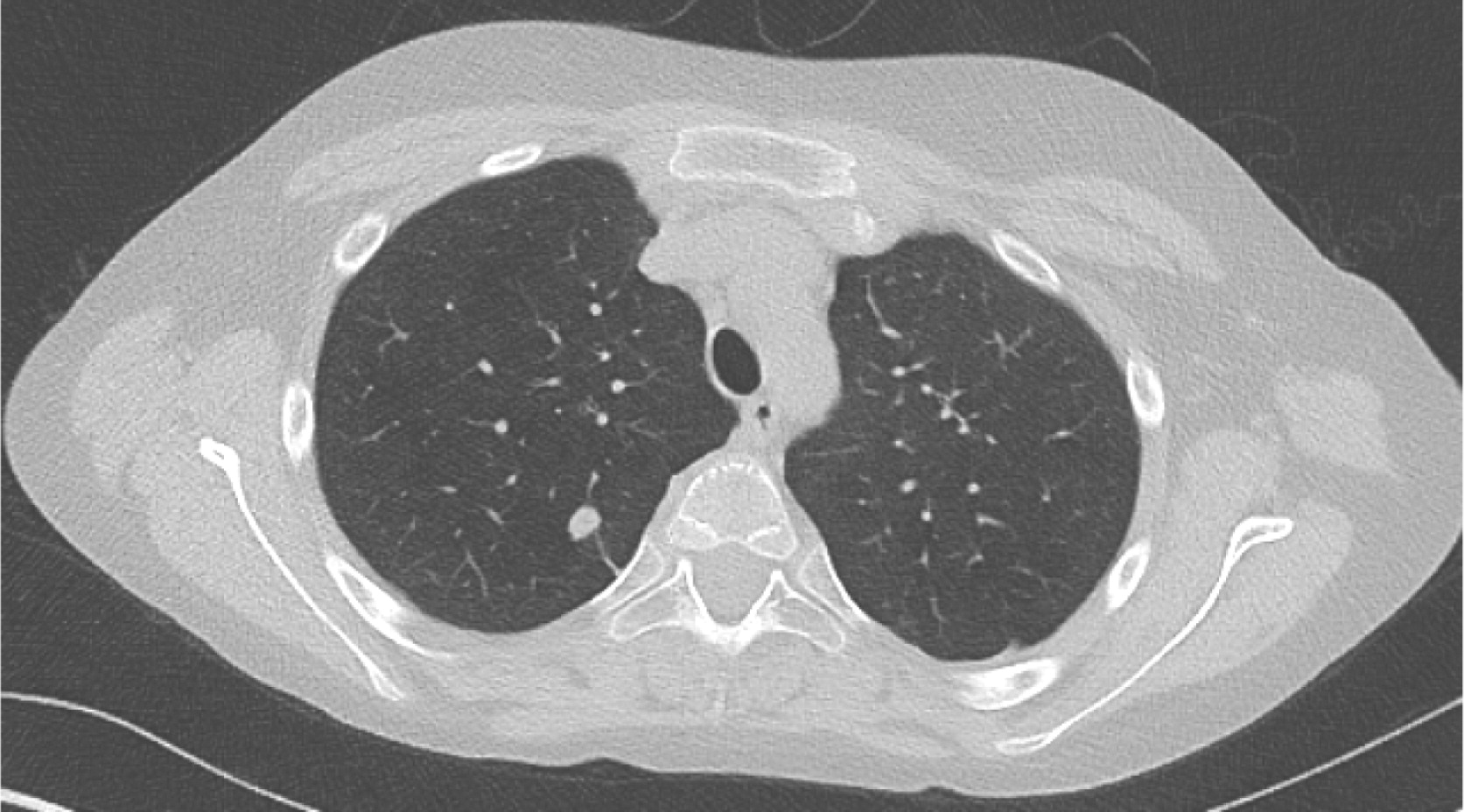
Patient 5 presented at 3 years of age with a history of persistent nonbilious vomiting for almost 2 weeks. Upper gastrointestinal (GI) endoscopies revealed diffuse inflammation from the esophagus to the proximal region of the small bowel. Biopsies revealed a focal eosinophilic abscess in the duodenum but no granulomas. He was given the diagnosis of eosinophilic gastroenteritis and was started on omeprazole and ketotifen. However, vomiting and weight loss persisted for the next 6 months. Repeat upper- and lower-GI endoscopies no longer showed eosinophilic gastroenteritis. Biopsies of the colon and rectum revealed PAS+ (Periodic acid-Shiff) staining of macrophages in the rectum that raised the possibility of CGD, a diagnosis which was later confirmed. Vomiting was explained by gastric outlet obstruction, a gastrojejunal tube was inserted for feeding, and he was started on steroids for the gastrointestinal inflammation. Prophylactic trimethoprim–sulfamethoxazole was also added. He had a human leukocyte antigen (HLA)-matched sibling; however, the family did not agree to proceed with HSCT. He was lost to follow-up for several years while he was treated persistently with steroids. He was admitted at the age of 12 years to the intensive care unit with septicaemia caused by small bowel microperforations (Figure 17) requiring small bowel resection and ileostomy. The resected portion of small bowel as well as biopsies from other GI sites showed nonnecrotizing granulomas. The procedure was later complicated with an abdominal abscess. Subsequent magnetic resonance imaging (MRI) of the pelvis revealed persistent bowel inflammation mimicking IBD (Figure 18). A CT scan of his chest showed multiple nodules/granulomas in his right lung (Figure 19).
Figure 17:
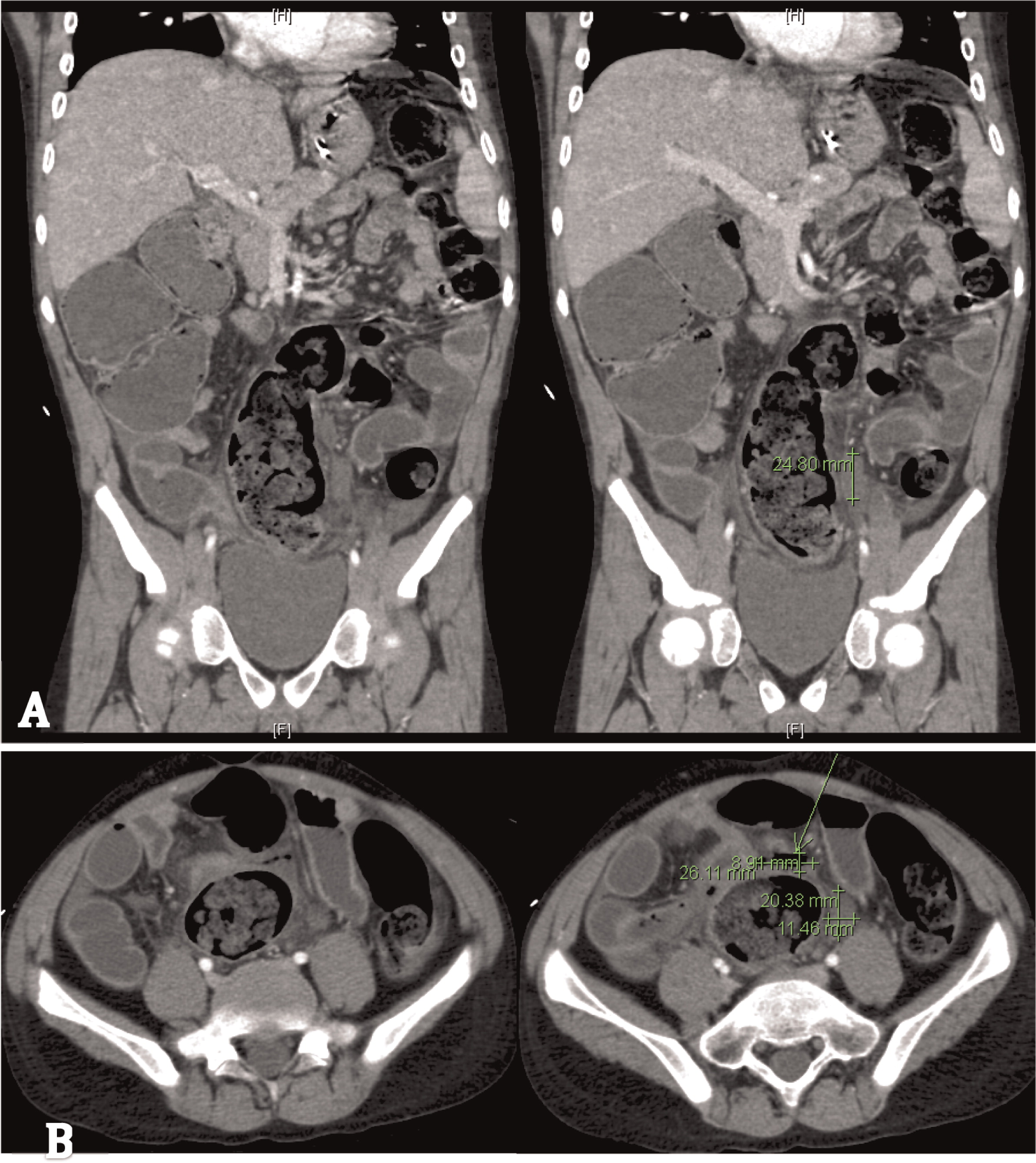
Figure 18:
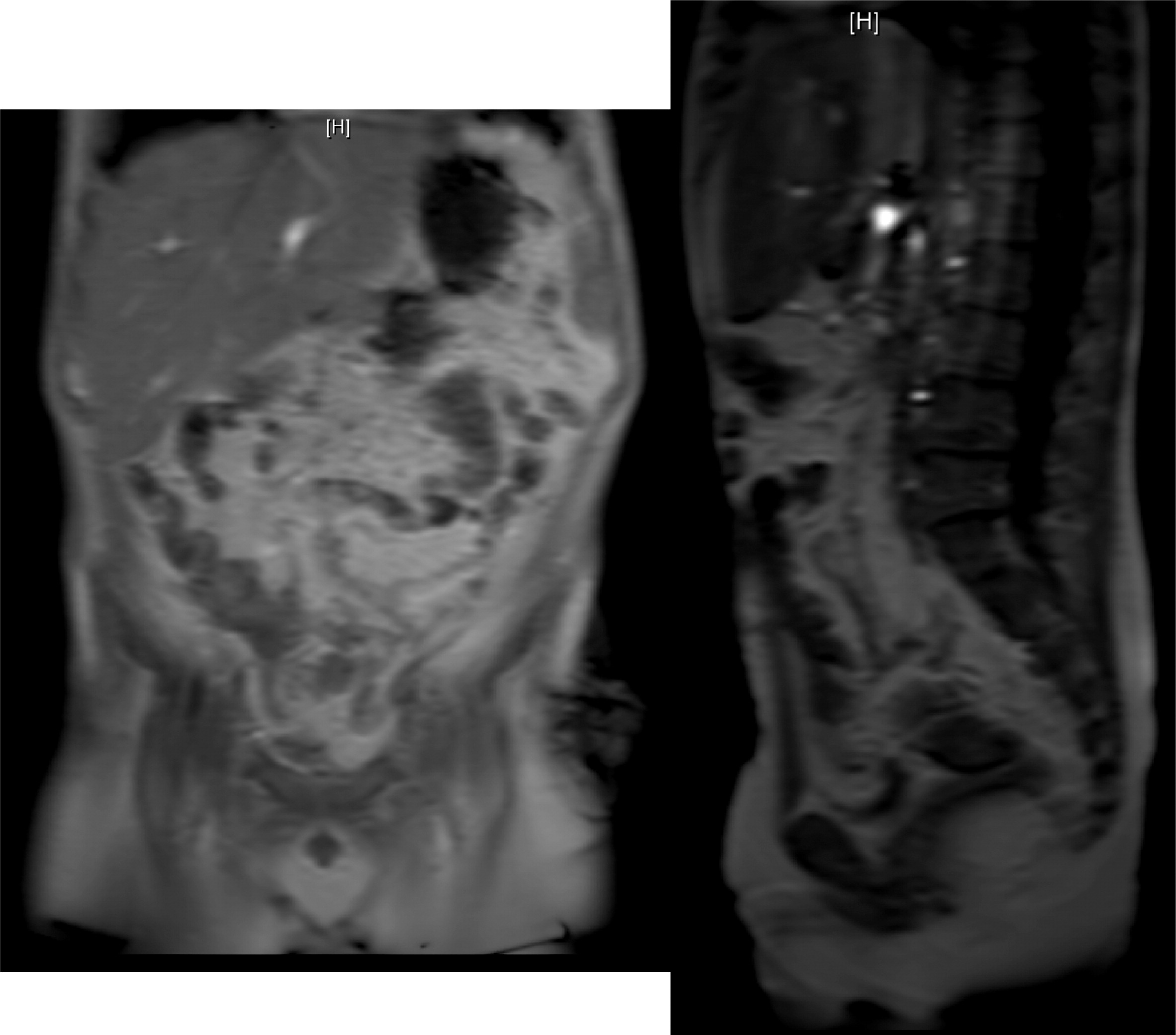
Figure 19:
Patient 6
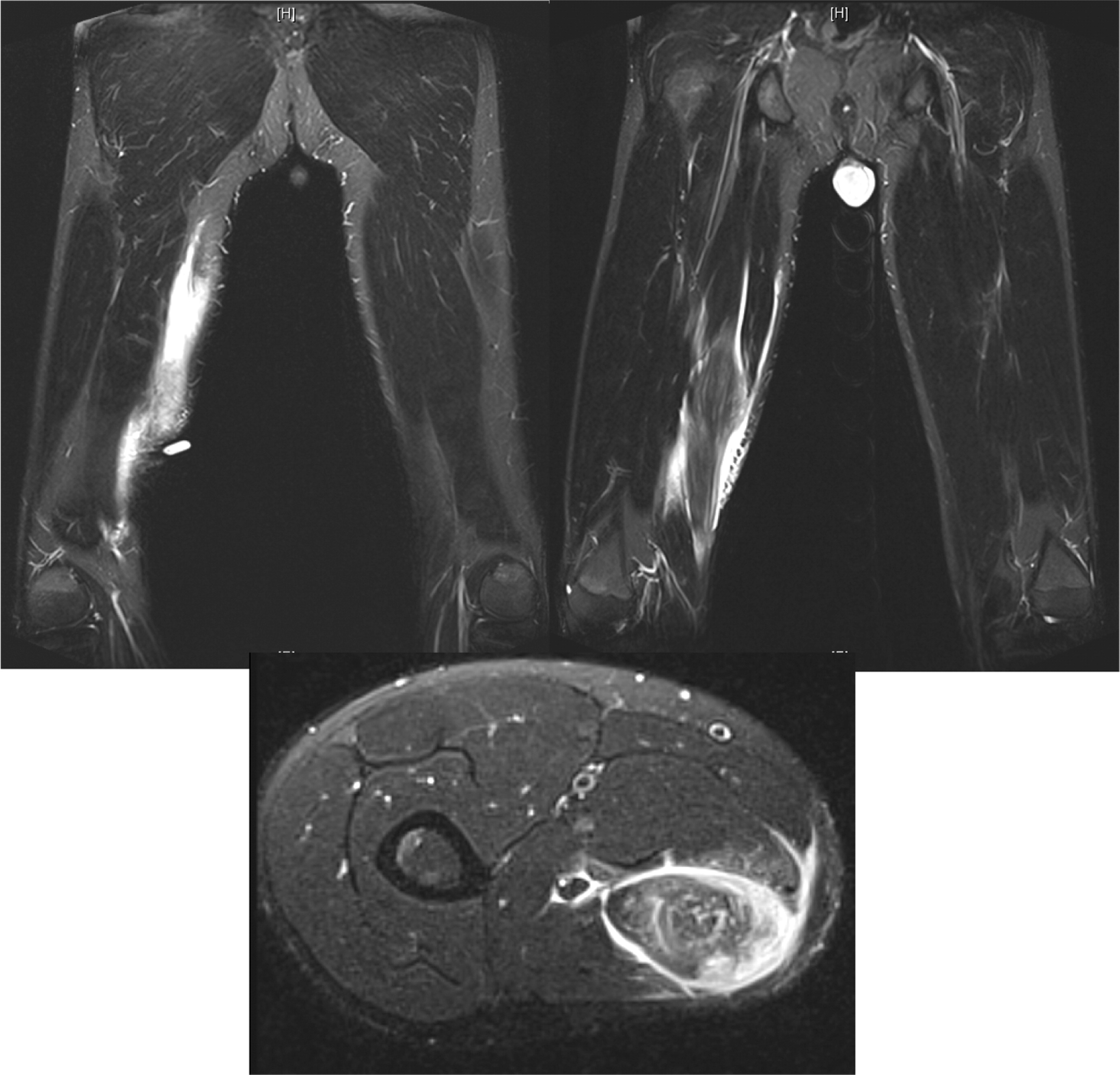
Patient 6 presented at 10 months of age with bilateral cervical lymphadenitis (Figure 20). Lymph node excisional biopsy revealed granulomatous inflammation with no organisms. X-linked CGD was subsequently diagnosed. He was started on prophylactic antibiotic, antifungal, and interferon gamma. By the age of 7 years he experienced recurrent mouth ulcers frequently interfering with food intake. He was also suffering from intermittent anal fissures and recurrent skin infections complicated with abscesses despite prophylactic trimethoprim–sulfamethoxazole and itraconazole as well as subcutaneous interferon gamma. The patient decided to discontinue interferon gamma at the age of 11 years and prophylactic antibiotic and antifungals at 14 years of age. At 15 years of age he was admitted with an abscess of his right foot, right thigh, and right lung. A CT scan of his lungs revealed an irregular shaped mass lesion involving the right middle and lower lung lobes and a small nodule in the left lower lung lobe (Figure 21). He also presented with extensive lymphadenopathy within the right hilum, posterior mediastinum, right paratracheal groove, aorto-pulmonary window, as well as left para-aortic region. A lung biopsy revealed Aspergillus. As for his lower extremity, an MRI showed an inflammatory/infective process in his right thigh (Figure 22). Skin biopsies from his right foot and from his right thigh revealed abscesses with granulomatous inflammation, but no microorganisms were found.
Figure 20:
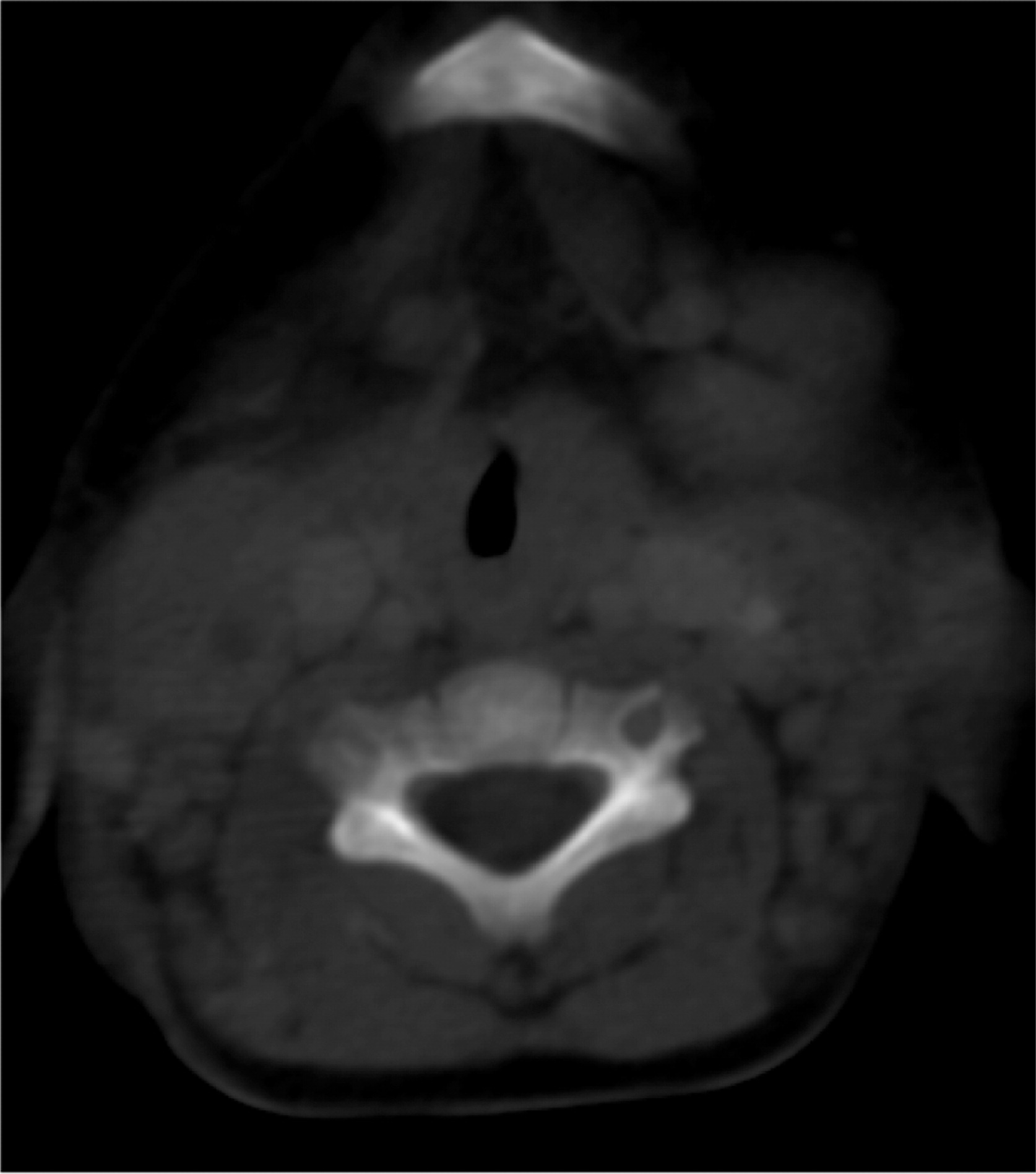
Figure 21:
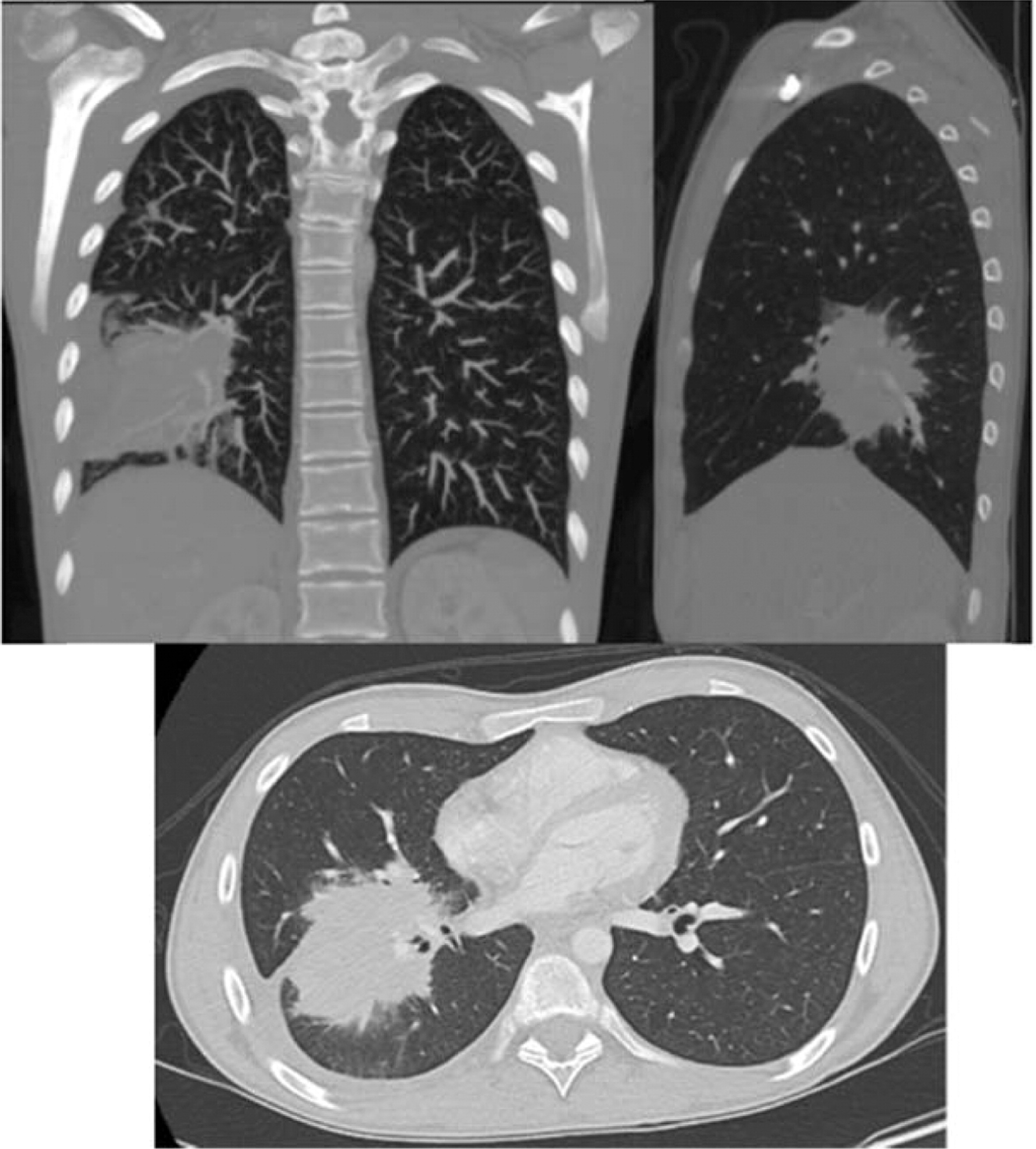
Figure 22:
Patient 7
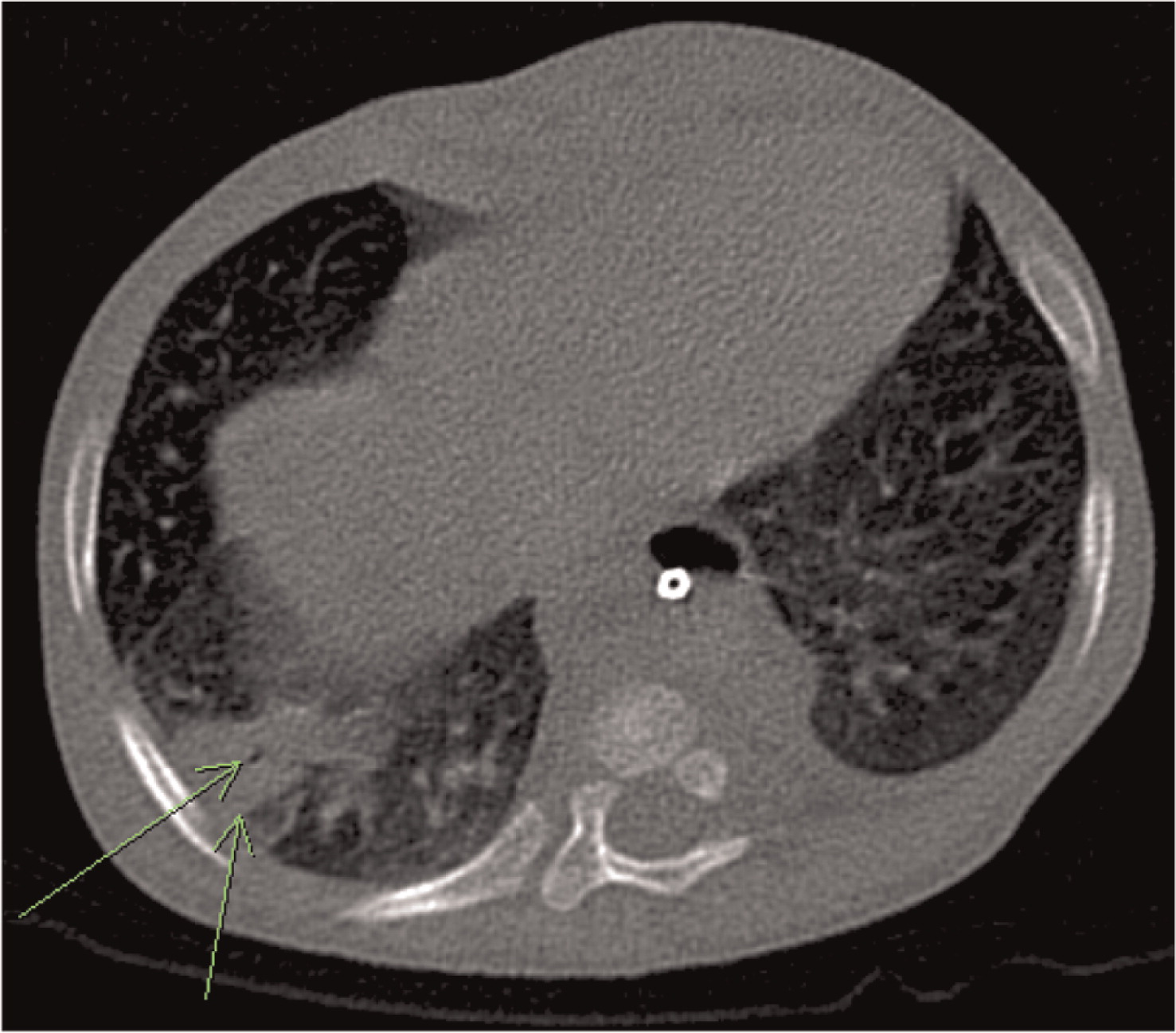
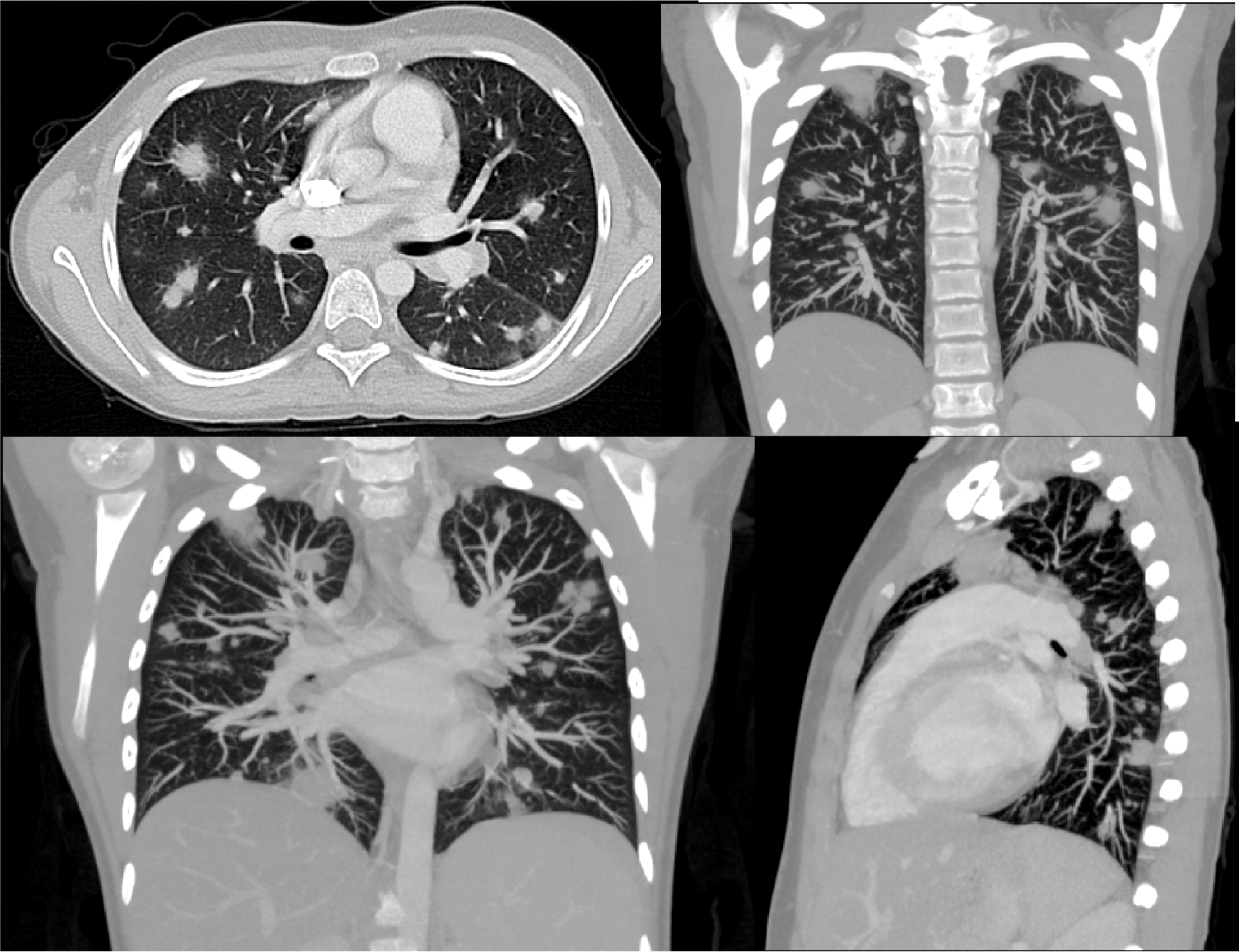
Patient 7 was a previously healthy male who presented at 12 years of age with sudden onset of swelling of many of his fingers and toes, daily fevers, mouth ulcers, decreased appetite, and weight loss. He was admitted to hospital, and the initial investigations showed signs of macrophage activation syndrome for which he was treated with high dose prednisone, and his symptoms progressively resolved. Two months later he was readmitted with fevers, painful oral ulcers, cough, and pleuritic chest pain. CT of his chest showed pulmonary nodules bilaterally with mediastinal lymphadenopathy (Figure 23). A broncho-alveolar lavage was positive for Aspergillus fumigatus. He was found to have a mutation in NCF1 gene encoding for the p47phox subunit of NADPH oxidase.
Figure 23:
Discussion
As evidenced by our patients, invasive bacterial and fungal infections are a significant threat for CGD patients. Radiological findings as well as the identification of the causative microorganisms, which often requires invasive procedures, are important diagnostic measures to guide optimal treatment (Towbin and Chaves 2010; Henriet et al. 2013).
Imaging of the lungs is very important as pulmonary manifestations are the most common features in CGD, presenting in as many as 80% of patients and representing the most important cause of death (Winkelstein et al. 2000). Chest radiographs usually show consolidations, reticulonodular opacities, and scarring. CT findings include consolidation, ground-glass opacities, tree-in-bud pattern, centrilobular or random nodules, bronchiectasis, septal thickening, air trapping, or scarring. Complications such as abscess formation, empyema, or lung fibrosis may also be found and are quite unique to CGD where contiguous spread to the chest wall and associated osteomyelitis of ribs and vertebrae may occur (Towbin and Chaves 2010; Mahdaviani et al. 2013).
More than half of the patients with CGD might show changes in the lymph nodes as evidenced by imaging. Most of these changes are due to suppurative adenitis. The cervical nodal chain is most frequently involved, although other nodal sites can be affected. On US, internal debris are usually present with compression of the infected lymph node. Unique features to CGD are the findings by CT of thick, enhanced septae and calcifications in more chronic disease. Nonsuppurative infection with granuloma formation can also occur in lymph nodes (Winkelstein et al. 2000; Towbin and Chaves 2010).
Multiple abscesses and (or) granulomas may be present in the liver of 25%–50% patients (Winkelstein et al. 2000; Towbin and Chaves 2010). Sporadic hepatic abscesses, without an inciting event such as trauma or systemic infection, are uncommon in children and thus, when found in a sporadic manner, they should raise the possibility of CGD. As shown in our patients, these lesions may also affect the spleen in up to one-third of patients. Interestingly, similar to the lungs, granulomas in the liver and spleen tend to wax and wane over time, and they generally disappear after treatment with HSCT.
As for the gastrointestinal tract, the most common imaging findings are bowel wall thickening mimicking IBD and thickening of the wall of the gastric antrum, the latter being a unique feature in CGD patients (Winkelstein et al. 2000; Towbin and Chaves 2010). Upper GI imaging studies can show gastric outlet obstruction with gastric dilatation, delayed gastric emptying, circumferential antral narrowing, and thickened gastric folds (Towbin and Chaves 2010).
Osteomyelitis in CGD is usually caused by either Serratia marcescens or Aspergillus and localized to the lower extremities, chest wall, and vertebrae (Winkelstein et al. 2000; Towbin and Chaves 2010).To our knowledge, muscle lesions such as the one shown here in patient 6 have not been previously reported.
This work illustrates how imaging studies are critical tools for diagnosis as well as management of patients with CGD.
Acknowledgements
This work was supported by Immunodeficiency Canada, the Canadian Center for Primary Immunodeficiency, the Program for Immunogenomics, and the Jeffrey Modell Foundation.
REFERENCES
Battersby A.C., Cale C.M., Goldblatt D., and Gennery A.R. Clinical manifestations of disease in X-linked carriers of chronic granulomatous disease J. Clin. Immunol. 2013 33 8 1276 -1284
Grez M., Reichenbach J., Schwäble J., Seger R., Dinauer M.C., and Thrasher A.J. Gene therapy for chronic granulomatous disease: the engraftment dilemma Mol. Ther. 2011 19 1 28 -35
Henriet S., Verwejj P.E., Holland S.M., and Warris A. Invasive fungal infections in patients with chronic granulomatous disease Adv. Exp. Med. Biol. 2013 764 27 -55
Holland S.M. Chronic granulomatous disease Hematol. Oncol. Clin. North. Am. 2013 27 1 89 -99
Kang E.M., Marciano B.E., DeRavin S.S., Zarember K., Holland S.M., and Malech H.L. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation J. Allergy. Clin. Immunol. 2011 127 6 1319 -1326
Mahdaviani S.A., Mohajerani S.A., Casanova J.L., Mansouri S.D., and Velayati A.A. Pulmonary manifestations of chronic granulomatous disease Expert. Rev. Clin. Immunol. 2013 9 2 153 -160
Rieber N., Hector A., Kuijpers T., Roos D., and Hartl D. Current concepts of hyperinflammation in chronic granulomatous disease Clin. Dev. Immunol. 2012 1 -6
Roos D. and de Boer M. Molecular diagnosis of chronic granulomatous disease Clin. Exp. Immunol. 2013 175 2 139 -149
Towbin A.J. and Chaves I. Chronic granulomatous disease Pediatr. Radiol. 2010 40 5 657 -668
Winkelstein J.A., Marino M.C., Johnston R.B. Jr, Boyle J., Curnutte J., Gallin J.I., Malech H.L., Holland S.M., Ochs H., Quie P., Buckley R., Foster C.B., Chanock S.J., and Dickler H. Chronic granulomatous disease: report on a national registry of 368 patients Medicine. 2000 79 3 155 -169
Information & Authors
Information
Published In

LymphoSign Journal
Volume 1 • Number 2 • December 2014
Pages: 105 - 120
History
Received: 2 October 2014
Accepted: 9 October 2014
Accepted manuscript online: 9 October 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Murguia-FavelaLuis and MansonDavid. 2014. Imaging in patients with chronic granulomatous disease. LymphoSign Journal.
1(2): 105-120. https://doi.org/10.14785/lpsn-2014-0020
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Radiographic features of patients with chronic granulomatous disease
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
