Lung histopathology evaluation of an X-MAID patient with a novel mutation in MSN
Abstract
Background: The cell cytoskeleton is regulated by the ezrin-radixin-moesin (ERM) family of proteins, forming links between transmembrane proteins and the underlying actin cytoskeleton. Phosphorylation and activation of these proteins enable interactions with partners critically involved in shape regulation, such as actin filaments, transmembrane proteins, and scaffolding proteins. The MSN gene encodes moesin, which is ubiquitously expressed in lungs, spleen, kidney, endothelial cells of vessels, lymphocytes, and neutrophils. Deficiency or dysregulation of moesin, called X-linked moesin-associated immunodeficiency (X-MAID), is characterized by severe leukopenia affecting T cells, B cells, and neutrophils. To date, the clinical picture of patients with X-MAID is variable.
Aim: We describe the presentation, immune-workup, and lung histopathology findings of a young male patient with X-MAID and multi-organ involvement, whose severe pulmonary vein stenosis necessitated a double lung transplant.
Methods: A thorough review of the patient’s chart was performed.
Results: The patient presented with a history of recurrent respiratory tract infections, oral thrush, and 3 major bacterial infections requiring admission and antibiotic therapy. His immune evaluation was remarkable for low T cells, and normal numbers of B and NK cells. At age 4 years he underwent a double lung transplant due to severe pulmonary vein stenosis and pulmonary hypertension. He further developed chronic kidney injury post-transplant. Clinical trio whole exome sequencing revealed a novel hemizygous variant in the MSN gene (c.278dupT; p.L93FfsX21), predicted to cause loss-of-function in moesin. Histologic evaluation of the lung tissue before transplantation identified profound abnormalities in alveoli formation.
Conclusion: Patients with moesin deficiency may present during infancy or childhood with a severe form of the disease, including combined immunodeficiency with lymphopenia and neutropenia, while adults may have a milder clinical picture. The novel MSN mutation described here adds to the known spectrum of disease and highlights the non-redundant functions of moesin, particularly in the lung.
Statement of Novelty: We report the first lung histopathological description of an X-MAID case, in a pediatric patient with recurrent infections, cytopenia, and autoimmunity who underwent a double lung transplant.
Introduction
The Ezrin-radixin-moesin (ERM) family of proteins are responsible for developing networks between transmembrane proteins and the actin cytoskeleton (Fehon et al. 2010). They are essential for normal membrane organization and function, preserving cell shape, microvilli, pseudopod/uropod and immune synapse formation, phagocytosis, and apoptosis, as well as signal transduction pathway control (Niggli and Rossy 2008).
All ERM proteins are characterized by a plasma membrane-associated N-terminal 4.1 ezrin, radixin, moesin (FERM) domain, followed by a long region with a high α-helical propensity, and a C-terminal domain (C-ERMAD: C-terminal ERM-association domain) which binds to the FERM domain, creating a head to tail folding structure (Fehon et al. 2010; Niggli and Rossy 2008) (Figure 1A). The FERM domain contains 3 subdomains: the F1 ubiquitin-like domain, F2 consisting of 4 α-helices, and F3 composed of a pleckstrin homology domain (Figure 1B). A preserved FERM domain is critical for the overall ERM protein function (Michie et al. 2019). Phosphorylation and subsequent activation of ERM proteins enable interactions necessary for shape regulation, including those of actin filaments, transmembrane proteins, and scaffolding proteins. The participation of phosphatidylinositol 4,5 biphosphate (PIP2) is required for ERM protein activation, by binding to the FERM domain and phosphorylating the C-ERMAD domain. (García-Ortiz and Serrador 2020).
Figure 1:
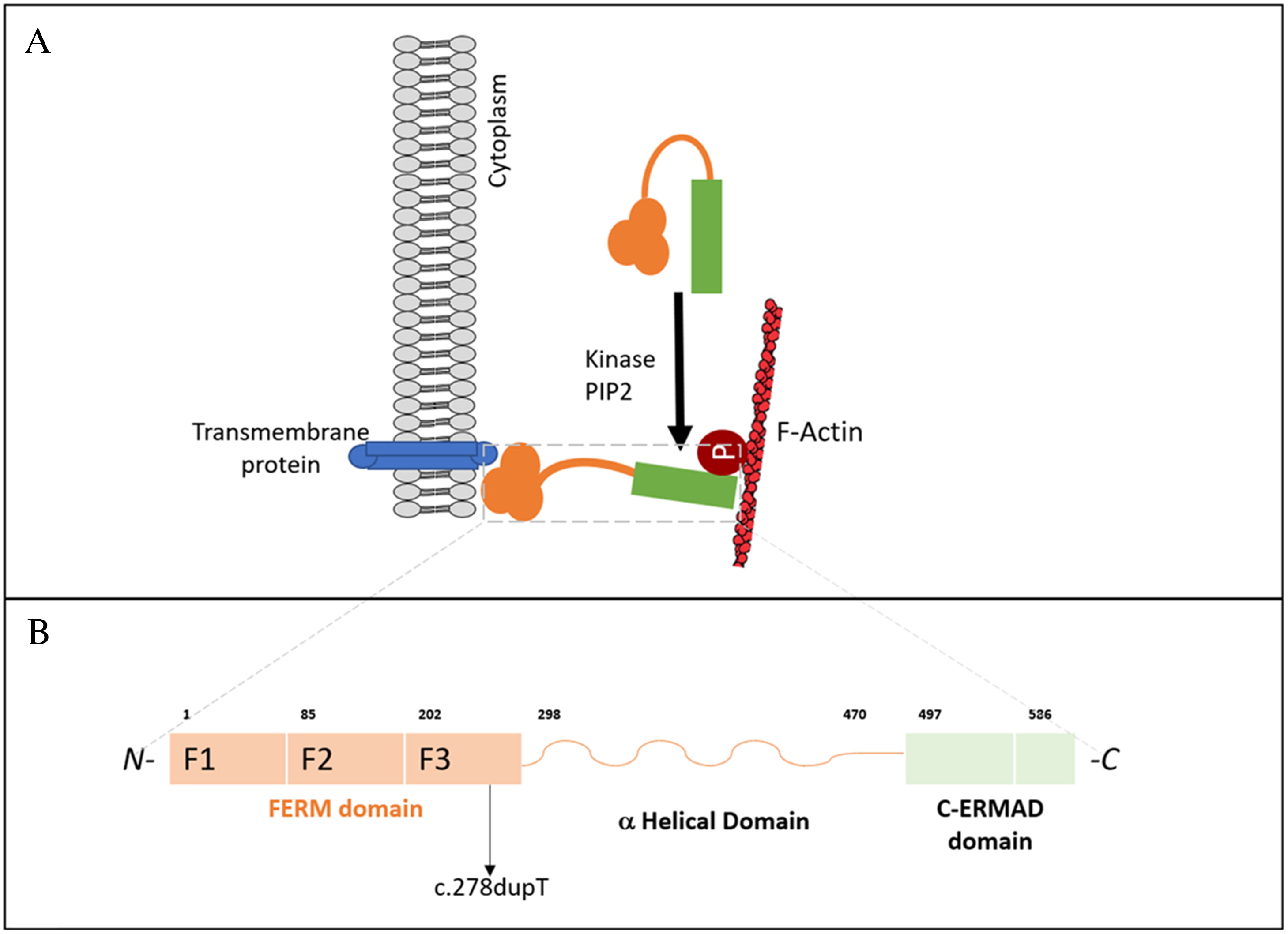
The MSN gene, located on the X-chromosome, encodes moesin which is ubiquitously expressed in lungs, spleen, kidney, endothelial cells of vessels, and is the predominant ERM protein in lymphocytes and neutrophils (Liu et al. 2015). Moesin is the abundant in leukocytes, while ezrin and radixin are near absent in leukocytes (García-Ortiz and Serrador 2020). Murine studies have previously demonstrated that moesin and radixin knockout mice appear clinically less severe. Mice reached adulthood despite abnormalities in the liver, lymphocytes and neutrophils, whereas knockout of ezrin resulted in embryonic lethality (Kawaguchi et al. 2017; Lagresle-Peyrou et al. 2016; Liu et al. 2015). Notably, loss-of-function of any one ERM protein is not compensated by other protein family members (Kawaguchi et al. 2017).
In recent years, a number of primary immunodeficiencies have been identified that result from aberrations in genes essential for actin cytoskeleton regulation. These fall under the scope of actinopathies, and affect cell motility, signaling, and antimicrobial function (Sprenkeler et al. 2021). X-linked moesin-associated immunodeficiency (X-MAID), first described in 2016, is part of a group of immunodeficiencies caused by defects in cytoskeleton. X-MAID is characterized by severe leukopenia affecting T cells, B cells, and neutrophils (Lagresle-Peyrou et al. 2016). To date, the clinical presentation of patients with X-MAID is variable, from those requiring minimal intervention, immunoglobulin replacement therapy and (or) antibiotic prophylaxis (Bradshaw et al. 2018; Henrickson et al. 2019; Lagresle-Peyrou et al. 2016), to patients needing life-saving hematopoietic stem cell transplant based on severe lymphopenia with significant clinical manifestations (Henrickson et al. 2019).
Here, we describe the presentation, immune-workup, and histopathology findings of a young male patient with X-MAID and multi-organ involvement, whose severe pulmonary vein stenosis necessitated a double lung transplant.
Methods
The patient was enrolled in the Primary Immunodeficiency Registry and Tissue Bank (REB protocol number 1000005598) at The Hospital for Sick Children. His chart was reviewed retrospectively.
Results
Case presentation
Our patient is currently a 9-year-old male from a non-consanguineous Caribbean family, born after an uncomplicated pregnancy at 38 weeks gestational age by emergency Cesarean section due to meconium. The neonatal period was remarkable for neonatal alloimmune thrombocytopenia, treated with intravenous immunoglobulins (IVIG). He had an episode of thrush around the age of 3 months which was treated with oral Nystatin. There were no other infectious or immune concerns in the first year of life.
At the age of 3 years, the patient presented to our Immunology Service with a history of recurrent infections, including affecting the recurrent respiratory tract as well as 3 major bacterial infections: Streptococcal bacteremia at age 2 years, group A Streptococcus pharyngitis, and periorbital cellulitis at around age 3 years, all requiring prolonged admission and intravenous antibiotic therapy. Additionally, he had high myopia, tortuous optic nerve identified on MRI, hypotonia, and motor developmental delay.
His immune evaluation revealed low CD3+ T cells, including both CD4+ and CD8+, and normal numbers of CD19+ and NK cells (Table 1). Other immune parameters were normal, including lymphocyte responses to phytohemagglutinin, as well as albumin and immunoglobulin levels, reactive specific antibody titers, neutrophil oxidative burst index, CH50, chromosomal microarray and chromosomal breakage assay.
Table 1:
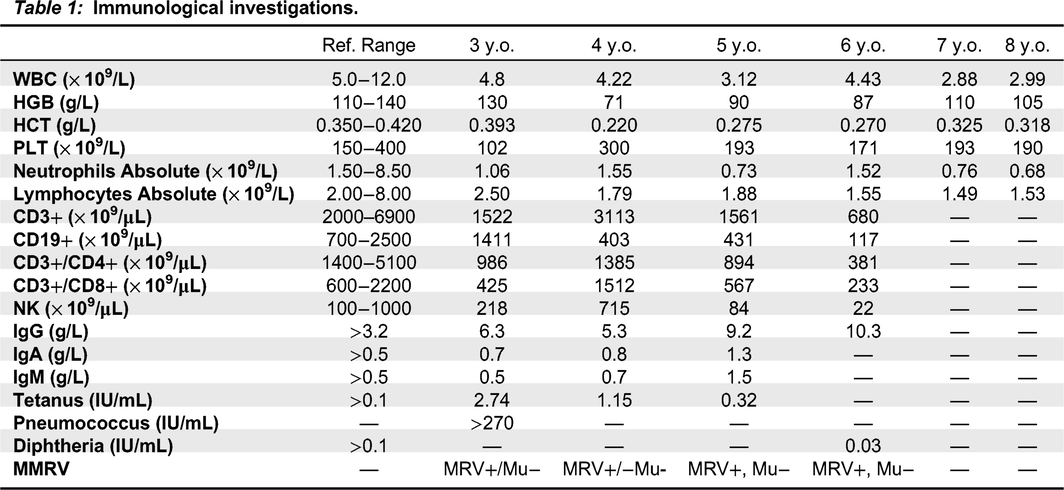
At age 4 years he was admitted for severe pulmonary vein stenosis, leading to pulmonary hypertension, and subsequently underwent a double lung transplant. In his pre-transplant workup, he was noted to have a positive Strongyloides serology, which was treated.
Post-transplant, he continued to suffer from multiple infections, including recurrent pneumonia (Staphylococcus, Pseudomonas, Hemophilus, Entero, Rhinovirus, MAC and Coronavirus), some requiring intravenous and in-hospital management. He had multiple hospital admissions due to hematemesis and bloody stools, with a gastrointestinal scope only showing mild gastritis and mildly prominent vessels in the esophagus and rectum. Further, he was unable to clear Norovirus from his GI tract.
Additionally, he developed immune-mediated anemia and neutropenia while he was on Tacrolimus and MMF. A prior bone marrow aspirate showed adequate granulopoiesis, as it was considered autoimmune, ultimately the cytopenia was managed with intravenous immunoglobulin and GCSF with a good response. A bone marrow aspirate and biopsy demonstrated normal granulopoiesis. In terms of other immune-mediated conditions, he also developed alopecia areata. He developed chronic kidney injury, which is believed to have resulted from immunosuppressive medications post-transplant.
Genetic findings
Clinical trio whole exome sequencing revealed a novel hemizygous variant in the MSN gene, encoding moesin (c.278dupT; p.L93FfsX21). Segregation analysis did not identify the variant in the patient’s mother or father, thus, it arose de-novo. Located in exon 4, the c.278dupT variant introduces a premature stop codon and is predicted to cause loss-of-function either through protein truncation or nonsense-mediated decay. Western blot evaluation confirmed reduced expression of moesin in the patient’s peripheral blood lymphocytes (not shown).
Lung histopathology analysis
Histologic evaluation of the lung tissue before transplantation (Figure 2) identified profound abnormalities in alveoli formation, showing enlargement of alveolar sacs, with emphysematous changes as compared to healthy control (Figure 2A, 2B). Also present are features of arteriolization of the small pulmonary veins in the lung parenchyma (see the venules that are indicated with short arrows in Figure 2B). Clinically, it increases flow resistance and attributes to the development of hypertension.
Figure 2:

Discussion
Moesin deficiency, also known as X-MAID has only recently been described. Severe forms of X-MAID may present during infancy or childhood with combined immunodeficiency (including lymphopenia and neutropenia), while adults appear to have a milder clinical picture (Lagresle-Peyrou et al. 2016). Other manifestations, including hypogammaglobulinemia and poor vaccine responses, have also been reported. With the recent introduction of newborn screening for severe combined immunodeficiency, cases of X-MAID have also been identified due to low levels of T cell receptor excision circles, demonstrating the importance of this screening tool (Delmonte et al. 2017; Dvorak et al. 2019).
The majority of previously reported MSN mutations affect the FERM domain, particularly the F2 subdomain (Bradshaw et al. 2018; Delmonte et al. 2017; Henrickson et al. 2019; Lagresle-Peyrou et al. 2016). Our patient’s de novo mutation similarly affected the FERM domain, but in the F3 subdomain (pleckstrin homology domain), essential for binding to transmembrane adhesion molecules. Aberrations this region, from amino acid 202 to 298, have not been extensively described. A recent case report of a de-novo MSN mutation affecting amino acid 217, was clinically described with some overlapping respiratory features like most X-MAID cases, but with marked features of autoimmunity and immune dysregulation (Kovács et al. 2022). Our patient also presented with autoimmune cytopenia and alopecia areata, which may represent a distinctive feature within these group of disorders. Moesin protein expression in lymphocytes was abnormal in our patient, similar to other case reports (Bradshaw et al. 2018; Lagresle-Peyrou et al. 2016).
Our histopathology evaluation of the lungs represent the first documented for a patient with X-MAID who suffered severe pulmonary vein stenosis, requiring a double lung transplant. The muscularization of the walls of the small pulmonary veins (so-called ‘arteriolization change’) results in sclerosis and loss of elasticity of the veins, causing functional obstruction of blood flow and hence hypertension. Moesin is expressed in high levels in the lung, indicating a critical role in the maintenance of alveolar structure as well as regulation of inflammation and repair. The absence of moesin expression likely explains the recurrent pulmonary disease since childhood in X-MAID patients. In line with our findings, the lungs of moesin-deficient mice demonstrate abnormal alveolar architecture with emphysematous changes, similar to what is seen in our patient. Further, in response to lung injury, moesin-deficient mice show greater lung damage and fibrosis compared to controls, indicating dysregulated repair mechanisms (Hashimoto et al. 2008). This is consistent with the high expression of moesin (compared to ezrin and radixin) in the lungs and its role in alveolar preservation and lung homeostasis (Hashimoto et al. 2008).
Taken together, the clinical presentation of our X-MAID patient described here adds to the known spectrum of disease and highlights the non-redundant functions of moesin, particularly in the lungs.
REFERENCES
Bradshaw G., Lualhati R.R., Albury C.L., Maksemous N., Roos-Araujo D., Smith R.A., Benton M.C., Eccles D.A., Lea R.A., Sutherland H.G., Haupt L.M., and Griffiths L.R. 2018. Exome sequencing diagnoses X-linked moesin-associated immunodeficiency in a primary immunodeficiency case. Front Immunol 9: 420.
Delmonte O.M., Biggs C.M., Hayward A., Comeau A.M., Kuehn H.S., Rosenzweig S.D., and Notarangelo L.D. 2017. First case of X-Linked Moesin deficiency identified after newborn screening for SCID. J. Clin. Immunol. 37(4): 336–338.
Dvorak C.C., Haddad E., Buckley R.H., Cowan M.J., Logan B., Griffith L.M., Kohn D.B., Pai S.-Y., Notarangelo L., Shearer W., Prockop S., Kapoor N., Heimall J., Chaudhury S., Shyr D., Chandra S., Cuvelier G., Moore T., Shenoy S., Goldman F., Smith A.R., Sunkersett G., Lugt M.V., Caywood E., Quigg T., Torgerson T., Chandrakasan S., Craddock J., Saldaña B.J.D., Gillio A., Shereck E., Aquino V., DeSantes K., Knutsen A., Thakar M., Yu L., and Puck J.M. 2019. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010-2018). J. Allergy Clin. Immunol. 143(1): 405–407.
Fehon R.G., McClatchey A.I., and Bretscher A. 2010. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11(4): 276–287.
García-Ortiz A. and Serrador J.M. 2020. Erm proteins at the crossroad of leukocyte polarization, migration and intercellular adhesion. Int. J. Mol. Sci. 21(4): 1502.
Hashimoto S., Amaya F., Matsuyama H., Ueno H., Kikuchi S., Tanaka M., Watanabe Y., Ebina M., Ishizaka A., Tsukita S., and Hashimoto S. 2008. Dysregulation of lung injury and repair in moesin-deficient mice treated with intratracheal bleomycin. Am. J. Physiol. Lung. Cell. Mol. Physiol. 295: 566–574.
Henrickson S.E., Andre-Schmutz I., Lagresle-Peyrou C., Deardorff M., Jyonouchi H., Neven B., Bunin N., and Heimall J. 2019. Hematopoietic stem cell transplant for the treatment of X-MAID. Front. Pediatr. 7: 170.
Kawaguchi K., Yoshida S., Hatano R., and Asano S. 2017. Pathophysiological Roles of Ezrin/Radixin/Moesin Proteins. Biol. Pharm. Bull. 40(4): 381–390.
Kovács A.L., Kárteszi J., Prohászka Z., Kalmár T., Késmárky G., Koltai K., Nagy Z., Sebők J., Vas T., Molnár K., Berki T., Böröcz K., Gyömörei C., Szalma J., Egyed M., Horváth S., Oláh P., Csuka D., Németh V., and Gyulai R. 2022. Hemizygous nonsense variant in the moesin gene (MSN) leads to a new autoimmune phenotype of Immunodeficiency 50. Front. Immunol. 13: 919411.
Lagresle-Peyrou C., Luce S., Ouchani F., Soheili T.S., Sadek H., Chouteau M., Durand A., Pic I., Majewski J., Brouzes C., Lambert N., Bohineust A., Verhoeyen E., Cosset F.L., Magerus-Chatinet A., Rieux-Laucat F., Gandemer V., Monnier D., Heijmans C., van Gijn M., Dalm V.A., Mahlaoui N., Stephan J.-L., Picard C., Durandy A., Kracker S., Hivroz C., Jabado N., Basile G.de S., Fischer A., Cavazzana M., and André-Schmutz I. 2016. X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. J. Allergy Clin. Immunol. 138(6): 1681–1689.e8.
Liu X., Yang T., Suzuki K., Tsukita S., Ishii M., Zhou S., Wang G., Cao L., Qian F., Taylor S., Oh M.J., Levitan I., Ye R.D., Carnegie G.K., Zhao Y., Malik A.B., and Xu J. 2015. Moesin and myosin phosphatase confine neutrophil orientation in a chemotactic gradient. J. Exp. Med. 212(2): 267–280.
Michie K.A., Bermeister A., Robertson N.O., Goodchild S.C., and Curmi P.M.G. 2019. Two Sides of the Coin: Ezrin/Radixin/Moesin and merlin control membrane structure and contact inhibition. Int. J. Mol. Sci. 20(8): 1996.
Niggli V. and Rossy J. 2008. Ezrin/radixin/moesin: Versatile controllers of signaling molecules and of the cortical cytoskeleton. Int. J. Biochem. Cell Biol. 40(3): 344–349.
Sprenkeler E.G.G., Webbers S.D.S., and Kuijpers T.W. 2021. When Actin is Not Actin’ like It should: A new category of distinct primary immunodeficiency disorders. J. Innate. Immun. 13(1): 3–25.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 10 • Number 1 • March 2023
Pages: 1 - 6
History
Received: 23 March 2023
Accepted: 27 March 2023
Accepted manuscript online: 29 March 2023
Version of record online: 29 March 2023
Copyright
© 2023.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
LauraAbrego Fuentes, Amarilla B.Mandola, and BoNgan. 2023. Lung histopathology evaluation of an X-MAID patient with a novel mutation in MSN. LymphoSign Journal.
10(1): 1-6. https://doi.org/10.14785/lymphosign-2023-0003
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Characterization of thymic architecture and lymphocyte populations in X-MAID due to an underlying pathogenic moesin mutation
