Novel heterozygous PIK3CD mutation presenting with only laboratory markers of combined immunodeficiency
Abstract
Introduction
Methods
Patients
Lymphocyte proliferation
Western blotting
Whole exome sequencing and variant calling
Sanger sequencing
Results
Patients
Immune evaluation
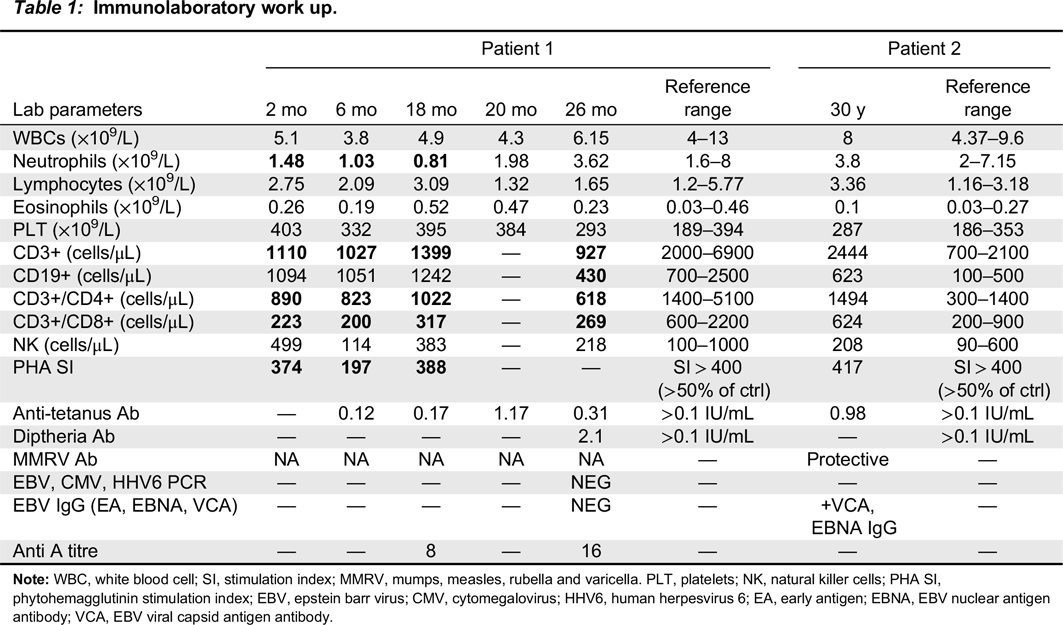
Note: WBC, white blood cell; SI, stimulation index; MMRV, mumps, measles, rubella and varicella. PLT, platelets; NK, natural killer cells; PHA SI, phytohemagglutinin stimulation index; EBV, epstein barr virus; CMV, cytomegalovirus; HHV6, human herpesvirus 6; EA, early antigen; EBNA, EBV nuclear antigen antibody; VCA, EBV viral capsid antigen antibody.
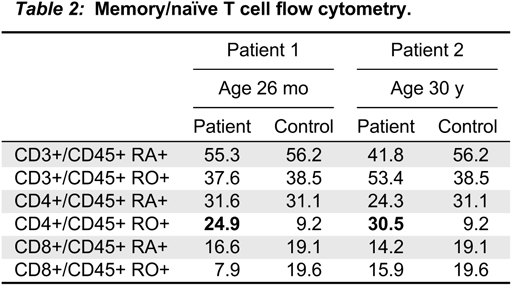

Note: NBS, newborn screening; TRECs, T cell receptor excision circles; WB, whole blood.
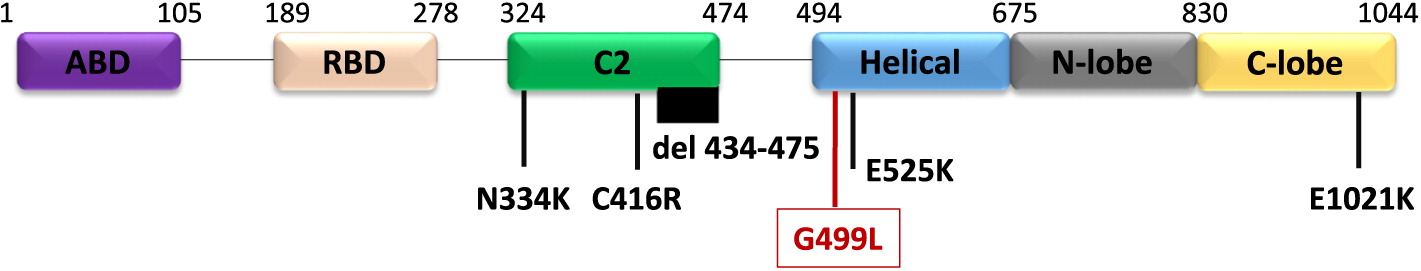
Genetic work up

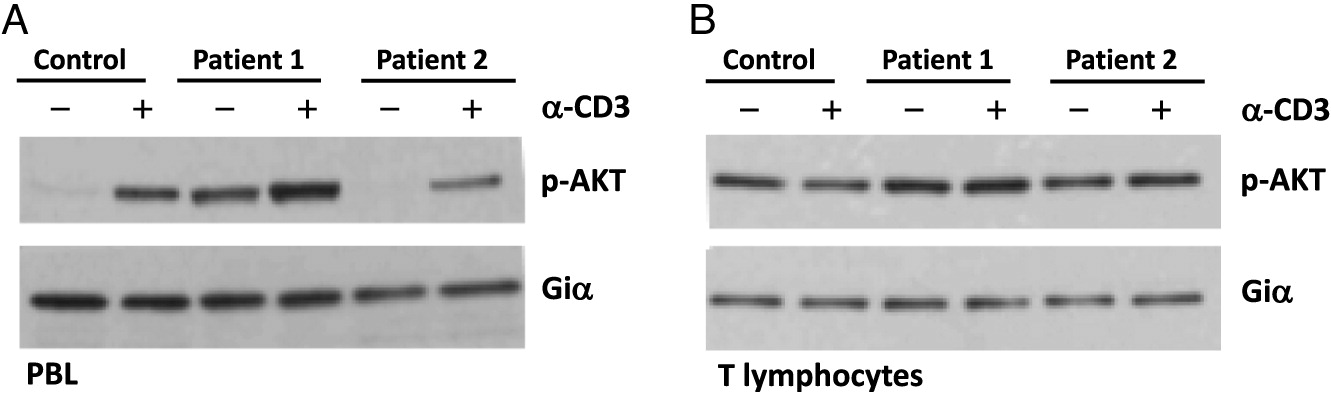
Cell signaling
Discussion

Conclusion
REFERENCES
Information & Authors
Information
Published In

History
Copyright
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
View Options
View options
Get Access
Login options
Check if you access through your login credentials or your institution to get full access on this article.
