An atypical presentation of ataxia telangiectasia in a school-aged boy secondary to an intronic mutation
Abstract
Background: Ataxia telangiectasia typically presents in early pre-school years with progressive cerebellar ataxia and oculocutaneous telangiectasias. Referral to Immunology is often made after diagnosis has been established, as patients are prone to both humoral and cellular immune abnormalities.
Case presentation: We herein report a 10-year old boy, previously undiagnosed, who presented with recurrent pneumonias. On history, frequent falls and speech difficulty were reported, with no telangiectasias on exam. Screening with alpha-fetoprotein was abnormal, followed by ATM gene sequencing, showing a homozygous intronic mutation. Over the next 3 years the patient deteriorated neurologically, and developed appreciable telangiectasias.
Conclusion: A review of the literature demonstrates that intronic/splicing mutations may result in atypical ataxia telangiectasia phenotypes and delayed presentations. We advise immunologists to have a high index of suspicion for ataxia telangiectasia when assessing a patient with immunodeficiency and neurologic regression, regardless of age, and even in the absence of telangiectasias.
Statement of novelty: We present a case of phenotypically atypical (“leaky”) ataxia telangiectasia resulting from a novel homozygous splice-site mutation in the ATM gene. Given high reported prevalence of intronic and splice-site mutations in ATM, we recommend full gene sequencing in patients suspected to have ataxia telangiectasia, especially in those with late onset or unusual manifestations.
Introduction
Ataxia telangiectasia (AT) is a DNA breakage syndrome caused by mutations in ATM, a large gene comprising of 66 exons as well as pseudo-exons (Platzer et al. 1997). The ATM gene product, the serine/threonine kinase ATM, is essential in induction of cell-cycle arrest and activation of repair mechanisms following detection of double-stranded DNA breaks. Given the ubiquitous nature of ATM activity, AT is marked by a multi-systemic presentation. A hallmark feature of AT is early-onset progressive ataxia, usually occurring between ages 2 and 5 years, with patients often becoming wheelchair-bound by age 10. Other typical manifestations include oculocutaneous telangiectasias, sinopulmonary infections, radiosensitivity and malignant predisposition (Teive et al. 2015). Surrogate markers in patients with AT include an elevated alpha fetoprotein (AFP), and abnormal chromosomal breakage studies. However, a definitive diagnosis requires the detection of biallelic mutations in ATM (Platzer et al. 1997; Yanofsky et al. 2009; Teive et al. 2015).
In recent years, cases of atypical AT have been increasingly reported. Such patients may present with unusual features such as dyskynesia, dysarthria, hypotonia, parkinsonism, or early malignancy, with few or no telangiectasias. While elevated AFP is seen in most cases, biallelic exonic mutation in ATM are not always detected (Platzer et al. 1997; Yanofsky et al. 2009; Teive et al. 2015). We hereby present a school-aged boy, who presented with late-onset progressive neurological decline and sinopulmonary infections. He was found to have a novel homozygous intronic mutation in the ATM gene, establishing a novel cause of atypical AT.
Functional and clinical presentation
Case presentation
A 10-year-old boy, recent migrant from Saudi Arabia, presented to our Immunology clinic for evaluation of recurrent sinopulmonary infections. These included a number of radiologically-confirmed pneumonias, acute otitis media, and 1 occasion of bacterial throat infection. All were treated successfully with oral antibiotics in his country of origin. He had never had any opportunistic, invasive or fungal infections, and no history of abscesses. Concurrently, the boy was complaining of frequent falls, as well as speech and fine motor difficulties. His parents described him as a “clumsy” child, with worsening gait instability in the preceding months. Moreover, the boy’s speech was becoming unclear, and he was struggling with fine motor tasks such as opening toothpaste tubes or turning door knobs. The boy also reported on fatiguability when trying to maintain an upright posture or stand for a prolonged period of time.
Past medical history included an unremarkable pregnancy and delivery. The neonatal period was only remarkable for jaundice, treated successfully with phototherapy. He had no other medical conditions or allergies, and was not taking any regular medications. He was fully immunized with all age-appropriate immunizations. On review of family history, parents were found to be first cousins of Pakistani origin, both with type 2 diabetes mellitus. A paternal aunt was reported to have been wheelchair-bound by age 20, and passed away at age 22. A maternal second cousin had never been ambulatory, and died at age 6. There was no known family history of any malignancies, including breast cancer.
On examination, the boy’s growth parameters and vital signs were within the normal range for this age. Cardiovascular, respiratory, abdominal and skin exam was normal. On neurological exam, the patient had evidence of horizontal nystagmus and dysarthria. He had mild hypotonia, diminished +1 reflexes in upper and lower limbs, and normal strength. Dysmetria and dysdiadochokinesia were noted, as well as mild intention tremor and difficulty with tandem gait, but no significant ataxia.
Investigations
Laboratory investigations showed a normal complete blood count and differential with the exception of mild lymphopenia. The white blood cell count was 8.6 × 109/L, hemoglobin 152 mg/L, platelets 239 × 109/L, neutrophils 6.09 × 109/L, lymphocytes 1.58 × 109/L, and monocytes 0.77 × 109/L. Lymphocyte subset analysis was normal apart from borderline low CD4 count. This included CD3 count of 1146 cells/μL (954–2332), CD8 of 500 cells/μL (282–749), CD4 of 607 cells/μL (610–1446), CD19 230 cells/μL (173–685), and NK 291 cells/μL (87–504). He had slightly elevated IgA of 3.1 g/L (0.5–2.2), but normal IgG of 9.5 g/L (6.6–15.3), and IgM of 1.5 g/L (0.4–1.5). He had appropriate vaccine responses to tetanus, diphtheria, measles, mumps, varicella, rubella. T-cell stimulation index to PHA was borderline low at 241 (normal: ≥300).
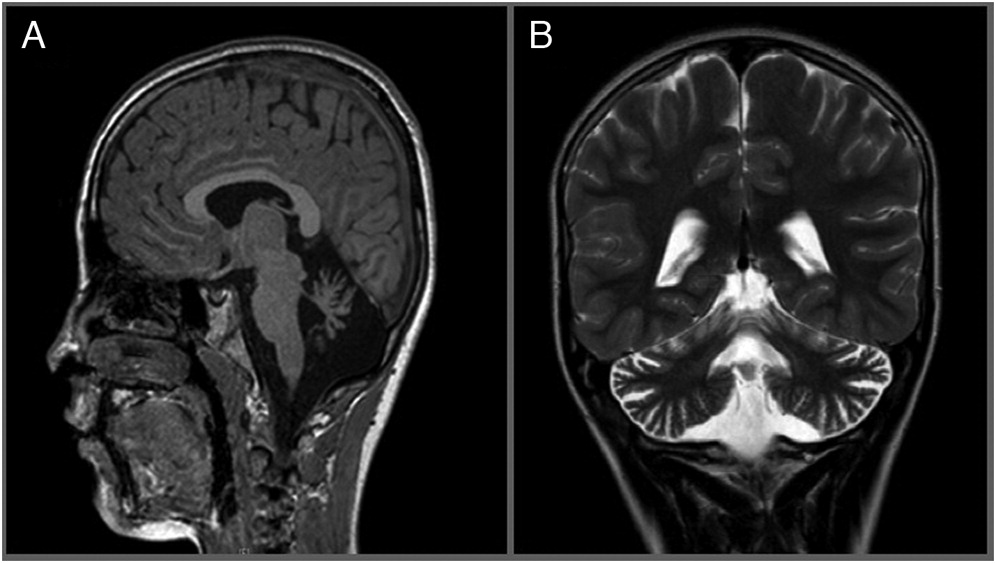
Brain magnetic resonance imaging (MRI) showed atrophy of the cerebellar vermis (Figure 1). This was followed by electromyography (EMG) which was unremarkable. Despite an unusual neurologic presentation and no evidence of telangiectasias at the time, the abnormal cerebellar findings on MRI raised suspicion for atypical ataxia telangiectasia, and prompted us to send an alpha fetoprotein level, which was grossly elevated at 155 μg/L (normal: 1–4). Targeted sequencing of the ATM gene was subsequently performed, revealing a novel homozygous intronic substitution mutation c.8269-7A>G in intron 56, in close proximity to an invariant splicing site.
Figure 1:
Outcome
In anticipation of challenges that the patient and family may encounter, and given the progressive nature of the condition, they have been connected with a multidisciplinary team, including neurology, ophthalmology, dermatology, occupational therapy, physiotherapy, and speech and language pathology. Given fatiguability, stooped posture and tremor, the patient was initially started on Carbidopa-Levodopa treatment, as benefit has been reported in some patients. Unfortunately, he experienced no improvement with this therapy. He was recently switched to Amantadine treatment. Over the 3 years that have elapsed since presentation, the patient continues to deteriorate. He has developed progressive ataxia, worsening dysarthria and intention tremor, and has become significantly fatigued even with standing. On recent exam, he now has visible telangiectasias on his sclerae and pinna.
Discussion
We report a case of a boy with late-onset AT and unusual disease manifestations, found to have a homozygous splice-site mutation in ATM. This may result in a “leaky” splicing or regulatory mutation, allowing for some gene product to be formed, possibly explaining the delayed presentation and unusual features. Our patient had 2 possibly affected cousins who had passed away at ages 6 and 22. Although the cause of death was never determined for those family members, it is likely that they had shared the same mutation as our patient, in particular given reported issues with ambulation in both cases. This suggests that prognosis may be similar for our patient, despite the relatively delayed onset of symptoms in this case.
The phenomenon of atypical or late-onset AT has been a topic of interest in recent years. In a study, Verhagen et al. (2012) demonstrated that the presence of residual ATM may result in a less severe phenotype. Cases correlating with residual kinase activity may manifest with less pronounced neurological symptoms, little or no telangiectasias, normal immunoglobulins levels, and later onset of malignancy (Verhagen et al. 2012). From a genetic standpoint, atypical or “leaky” presentations may be explained by regulatory or splicing mutations, allowing for some production of ATM kinase. Indeed, in a study, Teraoka et al. (1999) found that 30 out of 62 patients with ataxia telangiectasia had mutations resulting in defective splicing. Such mutations may result in exon skipping, intron retention, or activation of cryptic splice sites. Importantly, testing for ATM mutations in current clinical practice typically involves sequencing of the exonic portion of the gene only, which will not detect deep intronic mutations (Coutinho et al. 2005).
The notion of intronic splicing mutation in ataxia telangiectasia has other important implications which will need to be considered. From the prognostic perspective, data regarding long-term outcome in cases of atypical or leaky AT is scarce. A second point relates to family member counselling. It is known that heterozygous carriers of ATM gene mutations are at increased risk of breast cancer; however, it is unclear whether this is the case for intronic splicing mutations as well. In this regard, a recent study has identified intronic mutations resulting in micro-satellite instability in a subset of patients with gastric cancer, suggesting that such mutations may in fact be related to malignant predisposition (Kim et al. 2013). Finally, the presence of intronic mutations may have implications on developing therapies which target rescue of splicing defects, as has already been demonstrated both in vitro and in vivo for other conditions, such as Duchenne muscular dystrophy (Cavalieri et al. 2013).
We conclude that AT may present with unusual neurologic features, and with a late-onset. We caution clinicians to suspect a diagnosis of AT in patients with progressive cerebellar abnormalities and signs of immunodeficiency, even in the absence of telangiectasias and other classical features of AT. In such cases, screening with alpha fetoprotein is a simple, fast and reliable initial test, which should be elevated even in patients with atypical presentations. For definitive diagnosis, we recommend sequencing of the full ATM gene to detect deep intronic mutations. Further longitudinal data and research is required to counsel patients with atypical AT, as well as family members who are carriers of intronic mutations in the ATM gene.
REFERENCES
Cavalieri S., Pozzi E., Gatti R.A., and Brusco A. 2013. Deep-intronic ATM mutation detected by genomic resequencing and corrected in vitro by antisense morpholino oligonucleotide (AMO). Eur. J. Hum. Genet. 21(7): 774–778.
Coutinho G., Xie J., Du L., Brusco A., Krainer A.R., and Gatti R.A. 2005. Functional significance of a deep intronic mutation in the ATM gene and evidence for an alternative exon 28a. Hum. Mutat. 25(2): 118–124.
Kim H.S., Choi S.I., Min H.L., Kim M.A., and Kim W.H. 2013. Mutation at intronic repeats of the ataxia-telangiectasia mutated (ATM) gene and ATM protein loss in primary gastric cancer with microsatellite instability. PLoS ONE, 8(12): e82769.
Platzer M., Rotman G., Bauer D., Uziel T., Savitsky K., Bar-Shira A., Gilad S., Shiloh Y., and Rosenthal A. 1997. Ataxia-telangiectasia locus: Sequence analysis of 184 kb of human genomic DNA containing the entire ATM gene. Genome Res. 7(6): 592–605.
Teive H.A., Moro A., Moscovich M., Arruda W.O., Munhoz R.P., Raskin S., and Ashizawa T. 2015. Ataxia-telangiectasia—A historical review and a proposal for a new designation: ATM syndrome. J. Neurol. Sci. 355(1–2): 3–6.
Teraoka S.N., Telatar M., Becker-Catania S., Liang T., Önengüt S., Tolun A., Chessa L., Sanal Ö., Bernatowska E., Gatti R.A., and Concannon P. 1999. Splicing defects in the ataxia-telangiectasia gene, ATM: Underlying mutations and consequences. Am. J. Hum. Genet. 64(6): 1617–1631.
Verhagen M.M., Last J.I., Hogervorst F.B., Smeets D.F., Roeleveld N., Verheijen F., Catsman-Berrevoets C.E., Wulffraat N.M., Cobben J.M., Hiel J., and Brunt E.R. 2012. Presence of ATM protein and residual kinase activity correlates with the phenotype in ataxia-telangiectasia: A genotype–phenotype study. Hum. Mutat. 33(3): 561–571.
Yanofsky R.A., Seshia S.S., Dawson A.J., Stobart K., Greenberg C.R., Booth F.A., Prasad C., Del Bigio M.R., Wrogemann J.J., Fike F., and Gatti R.A. 2009. Ataxia-telangiectasia: Atypical presentation and toxicity of cancer treatment. Can. J. Neurol. Sci. 36(4): 462–467.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 7 • Number 2 • June 2020
Pages: 57 - 60
History
Received: 23 February 2020
Accepted: 18 April 2020
Accepted manuscript online: 4 May 2020
Copyright
© 2020.
Authors
Funding Information
This work has not been funded.
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
OriScott, YaelDinur-Schejter, JuliaUpton, and StephenFeanny. 2020. An atypical presentation of ataxia telangiectasia in a school-aged boy secondary to an intronic mutation. LymphoSign Journal.
7(2): 57-60. https://doi.org/10.14785/lymphosign-2020-0004
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Get Access
Login options
Check if you access through your login credentials or your institution to get full access on this article.
