Efficacy, safety, and tolerability of IVIG-SN in patients with primary immunodeficiency
Abstract
Background: IVIG-SN is a modern intravenous immunoglobulin with multiple pathogen elimination steps. This clinical trial in patients with primary immunodeficiency (PID) was designed to evaluate the safety, efficacy, and tolerability of this product in adults and children.
Methods: Forty-four patients with PID were treated with IVIG-SN for 12 months. IgG trough levels and pharmacokinetics of IVIG-SN were evaluated as well as efficacy and safety according to standard FDA guidelines.
Results: Overall, a total of 572 IVIG-SN infusions were administered according to either a 21- or a 28-day schedule. IgG trough levels during the treatment period ranged from 823.00 to 902.77 mg/dL, respectively, and half-life in serum of the administered IgG was 43.45 ± 30.25 days. There were no deaths and no adverse events leading to withdrawal from the study. Of all infusions administered, only 137 (24%) were temporally associated with an adverse event (AE). The upper bound for the 95% CI for the frequency of infusions temporally associated with an AE was 29.2%. Drug-related AEs were predominantly mild, and there were no acute serious bacterial infections during the study. Efficacy was also demonstrated by low rates of missed work, school, or daycare days (mean 2.6 days); unscheduled visits to physicians (mean 1.7); and therapeutic antibiotic use (mean 15 days).
Conclusion: IVIG-SN is effective in preventing infections and is safe and well tolerated.
Statement of novelty: This efficacy and toxicity trial was conducted using a new IVIG preparation.
Introduction
Purified IgG has been used for half a century as a therapeutic modality to replace antibody deficiencies. Immunoglobulin preparations suitable for intramuscular use were used for 30 years but were clearly sub-optimal, as trough levels were low and insufficient for prevention of major infections (Eibl 2008). The introduction of immunoglobulins suitable for intravenous use (IVIG) revolutionized treatment of these conditions. Roifman et al. (1987) demonstrated that a serum level of more than 500 mg/dL was superior to lower concentrations in preventing infections as well as improving lung disease. This threshold has been used for decades by the United States Food and Drug Administration (FDA) and health authorities worldwide as a minimum required trough level (Roifman et al. 1987; Eibl 2008; US FDA 2012; Guidelines for IVIG replacement 2014). Subsequently, several reports have demonstrated that higher trough levels (700–900 mg/dL) may have further benefits in protecting patients from infections. However, the optimal IgG trough level has never been established.
The clinical benefits of using IVIG preparations over intramusclar products includes the ability to administer larger doses of IgG in a short period of time, rapidly achieving the normal physiological concentrations with pharmacokinetic properties similar to native IgG (Roifman and Gelfand 1988).
Modern IVIG products have a good safety record and are manufactured using strict FDA guidance including incorporation of multiple effective measures of microbial inactivation (Hooper 2008; US FDA 2012). However, tolerability appears to be variable with some patients tolerating some products but not others. It is therefore important to introduce new IVIG products that provide diversity and favourable options for patients. Introduction of new safe products is also vital to generate a steady supply of a limited resource as well as to help keep product price affordable.
IVIG-SN is a modern era 5% liquid IVIG preparation produced by using Cohn–Oncley fractionization (Cohn et al. 1946; Melin et al. 1949) and DEAE-sepharose chromatography. Several measures have been employed for microbial inactivation including treatment with solvent detergent and nanofiltration. It is stabilized with maltose at 100 mg/mL.
We report here the results of a prospective multicentre clinical trial that studied the safety, efficacy, and tolerability of IVIG-SN in patients with primary immunodeficiency (PID).
Methods
Study product
The 5% liquid IVIG used in this study was manufactured by Green Cross, Korea, from U.S. harvested source plasma that was fully screened for blood borne pathogens. Cohn–Oncley fractionation technology was used followed by ion exchange chromatography (Cohn et al. 1946; Melin et al. 1949). Three virus removal and inactivation steps were employed including nanofiltration and treatment with both cold ethanol and solvent detergent. The final product is pH 4.4 with an osmolarity of 331 mOsmol/kg. IgA concentration is 10.8 µg/mL and there is 99.8% monomeric plus dimeric IgG. Anti-D was not detected, and anti-complimentary activity was 19.9%. The product contains substantial levels of anti-HBs, anti-measles, anti-diptheria, and anti-polio antibodies.
Study design
This was a prospective, open label, single-arm, multicentre, historically controlled, Phase III study measuring the safety, efficacy, pharmacokinetics, and tolerability of IVIG-SN in patients with PID, as defined by the International Union of Immunological Societies (IUIS) criteria (Geha and Notarangelo 2007). Uniquely, prior to study enrollment, confirmation of the PID diagnosis included a review of infection history, serum IgG concentration with no immunoglobulin therapy, and responses to vaccination with polysaccharide and protein vaccines, when available. In addition, a checklist of inclusion and exclusions criteria was completed by the investigators for each patient. The study design required at least 40 evaluable patients with well-defined PID.
Upon consenting and screening, eligible patients received intravenous infusions of the study product at the same dose and interval as used for their previous IVIG maintenance therapy. IVIG-SN was administered at a dose of 300–900 mg/kg (of body weight) every 21 or 28 days (± 4 days; depending on their pre-study IVIG treatment schedule) for a period of 12 months. The dose regimen remained unchanged throughout the study period unless there was a medically justified need to change it. The total patient follow-up was approximately 16 months during which patients underwent 13 treatment visits (patients on 28-day infusion schedule) or 17 treatment visits (patients on 21-day infusion schedule) and 2 follow-up visits. The first follow-up visit occurred 3 or 4 weeks after the last study drug infusion and the second follow-up visit (viral safety follow-up) was 3 months after the last study drug infusion.
During the treatment and follow-up periods, patients underwent the following evaluations: adverse event monitoring and recording of concomitant medications (throughout the study), review of patient diaries (at each treatment visit starting from the second infusion and at the first follow-up visit), physical exam (prior to each infusion and at the first follow-up visit), assessment of vital signs (multiple assessments at each visit including 10–15 min pre-infusion, 5 min before each increase of the infusion rate, 30 min after reaching the maximum infusion rate, every 60 min thereafter, and immediately after the infusion was completed), routine safety blood tests (hematology, blood chemistry), and urinalysis completed before each infusion and at the first follow-up visit. Measurements of direct anti-globulin (DAT/Coombs) were performed before and after the first infusion and before and after the 6th and 13th infusions (patients on 28-day infusion schedule) or before and after the 8th and 17th infusions (patients on 21-day infusion schedule). Viral safety testing was performed at week 12 (4th infusion) for patients on a 28-day infusion schedule or at week 15 (6th infusion) for patients on a 21-day infusion schedule. Viral safety testing was also performed at week 24 (7th infusion for patients on a 28-day infusion schedule and 9th infusion for patients on a 21-day infusion schedule), at the first follow-up visit (before the first administration of another IVIG product), and at the second follow-up visit (3 months after the last study infusion). Pharmacokinetic assessments were undertaken after the 5th study infusion and included 15 patients.
Summary of the inclusion criteria
Eligible patients were males or females aged 2 to 70 years, with PID (as defined by the IUIS criteria) who required treatment with IVIG. In addition to documented agammaglobulinemia or hypogammaglobulinemia, documented antibody deficiency was requested. Patients had to have received 300–900 mg/kg of a licensed IVIG therapy at 21- or 28-day intervals for at least 3 months prior to study enrollment and have had at least 2 documented IgG trough levels of ≥ 500 mg/dL, obtained at 2 scheduled infusions (21 or 28 days) within 12 months prior to study enrollment. Other IVIGs were prohibited 2 weeks prior to the first infusion of IVIG-SN and until the completion of the first follow-up visit. Patients who participated in a clinical trial of subcutaneous immunoglobulin (SCIG) were eligible as long as they switched to commercial IVIG for 3 infusion cycles (21 or 28 days) prior to enrollment in this study.
To participate in the study, all patients or their parents or guardians (for pediatric patients, defined as 2–17 years of age at study entry) had to provide written informed consent. In addition, pediatric patients required assent forms as appropriate per local regulations.
Summary of the main exclusion criteria
Patients were excluded from the study if they had any of the following: secondary immunodeficiency; newly diagnosed, previously untreated PID; dysgammaglobulinemia; isolated IgG subclass deficiency; history of repeated reactions or hypersensitivity to IVIG or other injectable forms of IgG; IgA deficiency with known antibodies to IgA; lifetime history of any thrombotic event; significant protein losing enteropathy, nephrotic syndrome, or lymphangiectasia; acute infection within 7 days prior to screening; known history or having tested positive at enrollment for human immunodeficiency virus (HIV) type 1 (by serology and NAT), HIV type 2 (by serology), hepatitis B virus (HBsAg and NAT), hepatitis C virus (by NAT), or hepatitis A virus (by NAT); profound anemia or persistent severe neutropenia; transaminases above 2.5 times the upper limit of normal, presence of a severe chronic disease, or other conditions precluding safe participation in the study. Other gender-specific exclusion criteria included current or planned pregnancy, breastfeeding, and lack of or refusal to use adequate contraception for women of childbearing potential; pregnancy was ruled out by a urine pregnancy test at screening.
Evaluation of pharmacokinetics
Pharmacokinetic (PK) evaluation was performed at or after infusion 5 of the study to ensure residual previous IVIG products had been washed out. Blood samples were taken before the infusion and at 15 min, 1 h, 24 h, 3 days, 7 days, 14 days, 21 days, and 28 days (for patients in the 28-day interval) during or after termination of the study. Samples were tested for the concentration of total IgG, IgG subclasses, and specific antibodies against tetanus toxoid, Streptococcus pneumoniae, and cytomegalovirus. Pharmacokinetic calculations included maximum serum concentration (C max), the time to reach maximum concentration (T max), IgG elimination half-life (T ½), the area under the concentration–time curve over one dosing interval (AUC0–t ), total body clearance, and volume of distribution (V 2).
Results
Patient demographics
Forty-four patients were enrolled to receive IVIG-SN. All patients had documented antibody deficiency and carried the diagnosis of either agammaglobulinemia or common variable immunodeficiency (Table 1).
Table 1:

Prior to enrollment, all patients were prescreened and were found to have hypogammaglobulinemia or agammaglobulinemia as well as a lack of specific antibodies. Investigators used a checklist to verify that all patients strictly met inclusion and exclusion criteria.
Patients were enrolled by 11 investigators (9 in the United States and 2 in Canada) and all 11 sites were activated following Institutional Review Board / Research Ethics Board approval. The patient profiles are as follows: 89% were Caucasian, 5% were African–American, 5% were Asian, and 2% were of undetermined race; 59% were male and 41% were female; mean age was 42 years; and 7 of the 44 patients were in the pediatric age group between 3 and 17 years of age (Table 1).
Infusion schedule
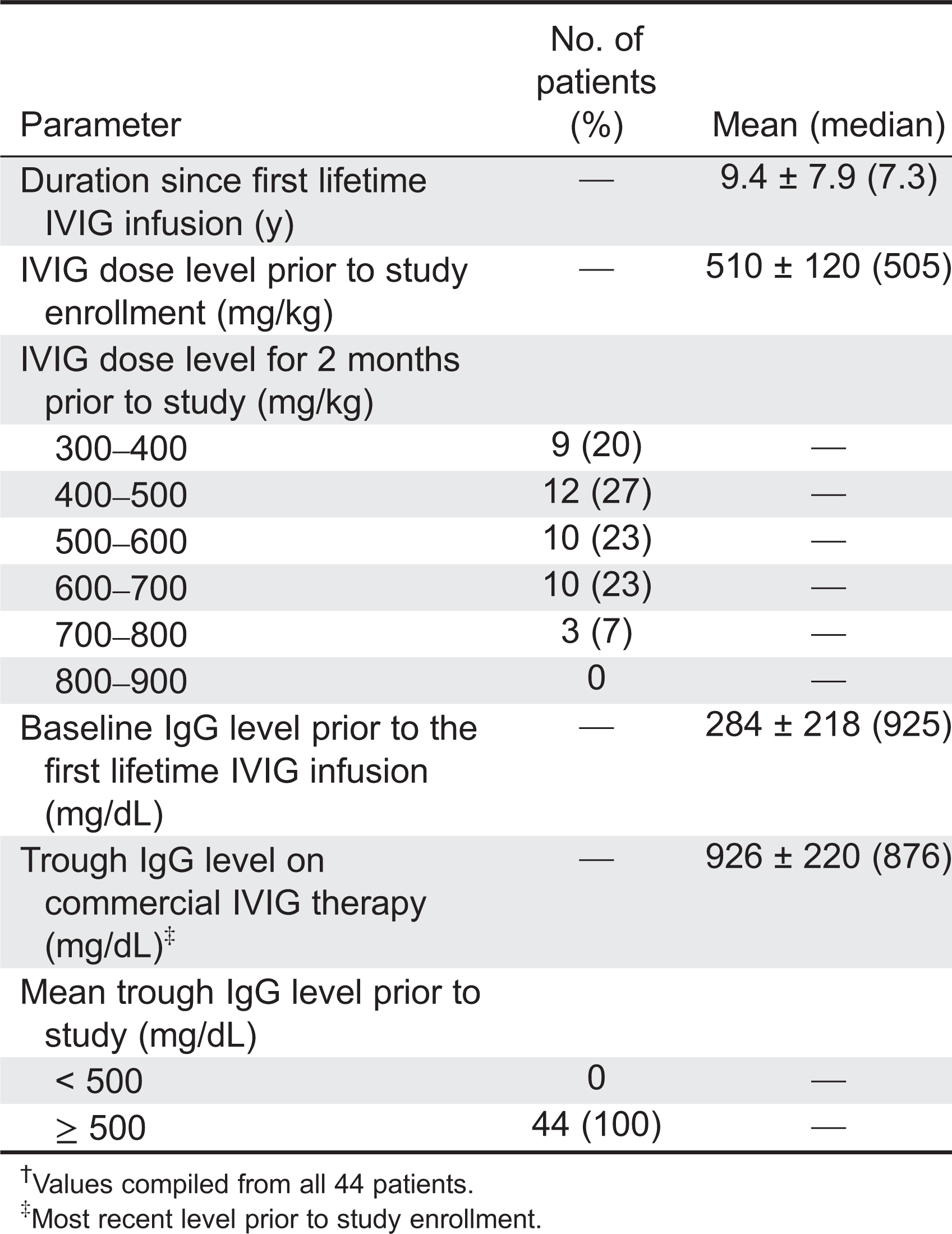
Dosing intervals were 21 days in 6 patients and 28 days in 38 patients. All 6 patients (100%) on the 21-day interval and 35 of 38 patients (92%) on the 28-day schedule completed the study. Historical trough levels prior to first replacement therapy were abnormal with a mean of 284 ± 218.70 mg/dL and a median of 203 mg/dL (Table 2). For the 12 months prior to study entry, IgG trough levels in all patients exceeded 500 mg/dL during replacement therapy. The mean IgG trough level prior to study initiation was 925.70 ± 220.02 mg/dL with a median of 876 mg/dL during IVIG treatment with doses ranging from 300 to 900 mg/kg with a mean of 510 ± 120 mg/kg and median of 505 mg/kg. During the study the mean dose administered was 515.4 ± 125.77 mg/dL with a median of 520 mg/dL, and the mean IgG trough level was almost identical for the 21- or 28-day schedules at 936.7 mg/dL and 908.4 mg/dL, respectively.
Table 2:
Efficacy of IVIG-SN
The primary efficacy endpoint of the study was the incidence of acute serious bacterial infections (meeting FDA criteria for bacterial pneumonia, bacteria/sepsis, osteomyelitis/septic arthritis, bacterial meningitis, or visceral abscess) per patient during the 12-month study period. A formal statistical analysis (noninferiority test) was applied for the occurrence of acute serious bacterial infection per patient. An upper 99% one-sided confidence limit of less than 1 per patient per year for the incidence of acute serious bacterial infection was predefined as an indicator of acceptable efficacy.
No acute serious bacterial infections occurred during the study, thus the efficacy of IVIG-SN was successfully demonstrated (upper limit of 99%, CI 0.00) (Table 3).
Table 3:

Efficacy was also demonstrated using secondary endpoints during the 1-year study period including:
(1)
The rate of missed work or school days were low (mean 2.6 days).
(2)
There were few unscheduled visits to physicians (mean 1.7 visits).
(3)
There was limited use of antibiotics (mean 15 days) (Table 3).
(4)
Three patients required hospitalization owing to an infection and the durations of these hospitalizations were 2–8 days (mean 0.4 days). In the overall study population, the mean annualized hospitalization rate was 0.1 days and the mean annualized duration of the hospitalizations was 0.5 days (Table 3).
(5)
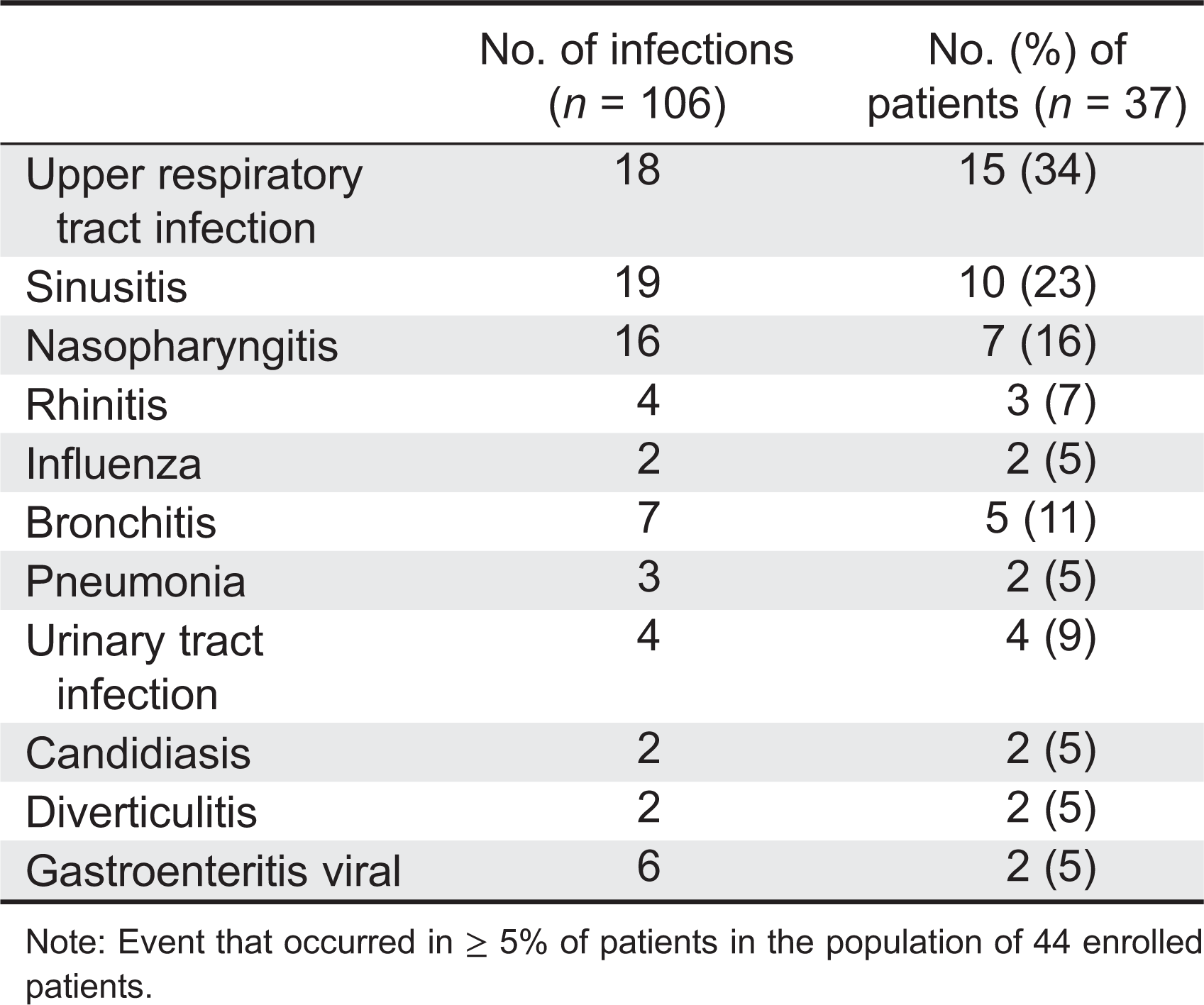
The total number of infections that occurred in 37 (84%) patients was 106, and most of these were minor. The majority of infections were unrelated: nasopharyngitis, sinusitis, and rhinitis represented 58 infections that occurred in 66% of the patients; 7 episodes of bronchitis were recorded in 5 patients; and there were 3 episodes of pneumonia (Table 4).
Table 4:
Adverse events
A total of 629 adverse events (AEs) were reported by the 44 patients. The majority of the AEs were defined as mild (79%) (Figure 1). Twenty-three events (4%), occurring in 9 patients, were designated as severe events. The majority of the AEs (473/629 or 75%) were not related to the study drug. Of the 629 AEs, 226 (36%) were temporally associated with the IVIG-SN infusion (occurring during or within 72 h of infusion), and the majority of these events occurred within the first 24 h after the infusion, whereas AEs occurred rarely during the infusion (Table 5). Common AEs occurring in ≥10% of the patients included: fatigue (32%); headache (30%); direct Coombs test positive (30%); upper respiratory infection (27%); nausea (27%); pyrexia (25%); diarrhea (23%); sinusitis (20%); cough (18%); vomiting (18%); abdominal pain (16%); pharyngitis, pain, nasal congestion, dizziness (14% each); and bronchitis, confusion, and myalgia (11% each).
Figure 1:

Table 5:
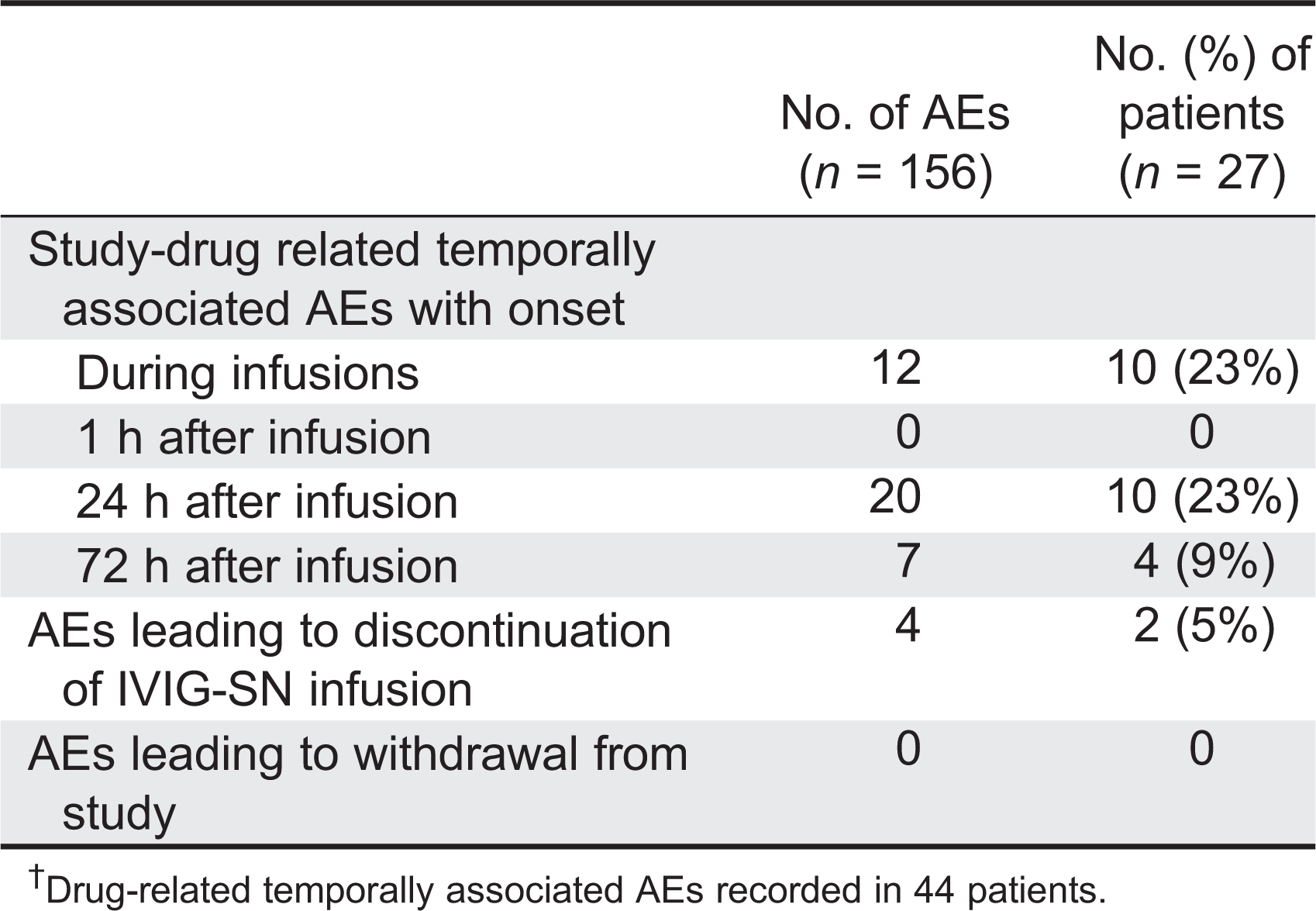
Overall, of the 572 infusions, 137 (24%) were temporally associated with an AE. The upper bound for the 95% CI for the frequency of infusions temporally associated with an AE was 29.2%. Therefore, we met the primary safety objective of demonstrating that less than 40% of infusions had 1 or more infusion-related AE.
Of the total of 629 AEs, the investigators assessed only 156 (25%) as related to the study drug. The most commonly reported treatment-related AEs were fatigue and positive Coombs test (25% each), headache (23%), and nausea (14%). Only 8 patients experienced treatment-related AEs that were assessed as certainly related to study treatment including: positive Coombs test (6 patients); headache and myalgia (2 patients each); fatigue, infusion site extravasation, and infusion site hematoma (1 patient). No deaths occurred during the study period.
A total of 11 serious AEs occurred in 6 patients during the study period. None was assessed as related to the treatment and none resulted in withdrawal from the study. Two patients had psychiatric disorders. Both of those patients had depression and one had drug dependency. Two patients had pneumonia; one with Pneumocystis jiroveci and one with unspecified pneumonia. Colitis, chest pain, spinal fracture, and renal failure appeared once.
Laboratory features and vital signs
Evaluation of laboratory tests did not indicate any safety concern for patients receiving IVIG-SN. No clinically meaningful changes were detected in hematology, clinical chemistry, vital signs, or weight measurements. Positive direct Coombs test was detected at least once during the study in 19 (43%) patients (2 of the 19 tested positive prior to exposure to the study drug). None of the 19 patients had any laboratory or clinical manifestation suggestive of hemolysis. One patient had a single positive HCV NAT test; however, serology and repeat HCV NAT were negative. One patient had transient HBV NAT positive. All other viral safety indicators including HIV1/HIV2 NAT and serology, HBV NAT and serology, HAV NAT and serology, and parvovirus B19 NAT and serology were negative, indicating lack of viral transmission through IVIG-SN treatment. Transient events of leukocytosis (1 patient), elevated liver enzymes (3 patients), and transient elevated creatinine unrelated to study drug and blood glucose levels (1patient each) were recorded.
In addition, blood pressure elevation was recorded in 1 patient and abnormal weight gain in another.
Pharmacokinetics
The PK population included 15 patients; 4 were on a 21-day schedule and 11 were treated every 28 days. PK parameters for total IgG are shown in the Table 6. The calculated serum half-life (T½, mean ± SD) was 25.53 ± 17.03 days for the 21-day patients, 49.97 ± 31.91 days for the 28-day patients, and 43.45 ± 30.25 days for all PK patients. Their T½, values corresponded to clearance rates. Mean volume of distribution (V2) ranged from 30 to 33 mL/kg and were similar to serum volume, implying minimal distribution into tissues. The median Tmax was 3.2–3.3 h from the initiation of the infusions with a mean infusion time of 2.48 h.
Table 6:
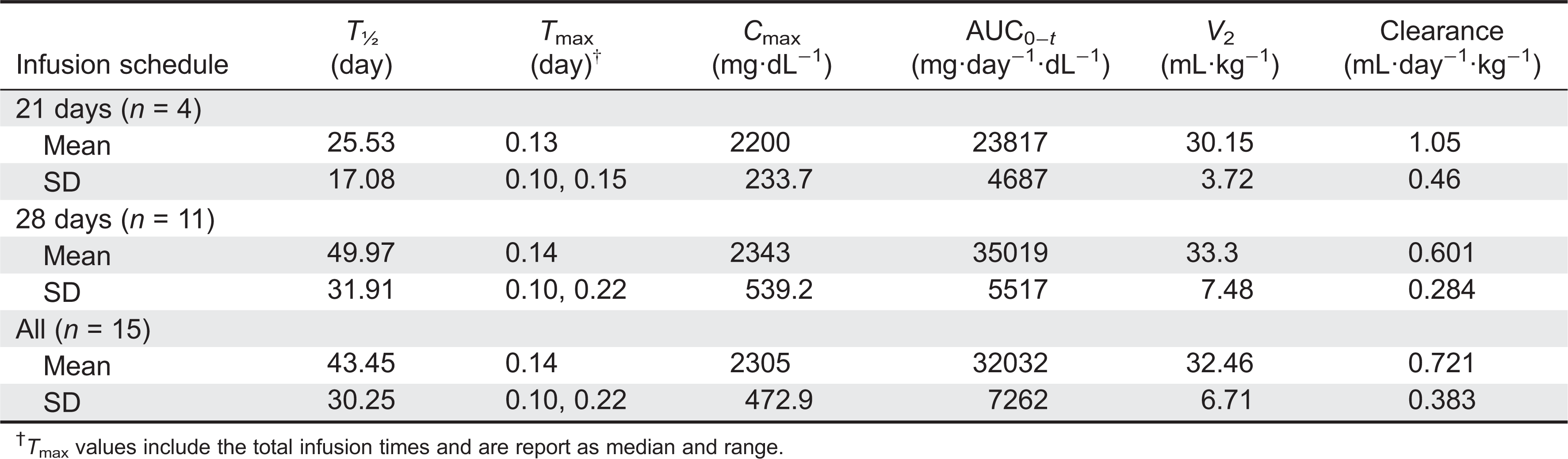
Mean Cmax was 2200 ± 233.7 mg/dL for 21-day schedule patients and 2343 ± 539.2 mg/dL for 28-day patients. Mean AUC0-t was 23 817 ± 4687 mg·day−1·dL−1 for 21-day patients, 35 019 ± 5517 mg·day−1·dL−1 for 28-day patients, and 32 032 ± 7262 mg·day−1·dL−1 for all PK patients.
For the IgG subclasses and specific antibodies, the mean T½ values were generally the same as for total IgG and illustrated that these analyses were not affected by the manufacturing process.
Discussion
This clinical trial was conducted to evaluate the safety, efficacy, tolerability, and bioavailability of IVIG-SN in patients with primary immunodeficiency. All updated FDA guidelines for IVIG studies were followed (US FDA 2012).
IVIG-SN was well tolerated by patients, as the rate of treatment-related AEs was low and the majority of reactions were mild. Changes in vital signs and body weight were also unremarkable in all but 2 patients. Laboratory tests were also completely normal for possible viral transmission in all cases, with occasional and temporary fluctuations in liver enzymes and blood count in only 2 patients. Direct Coombs test was positive periodically but it had no clinical significance, as observed in other clinical trials of IVIG (Roifman et al. 2003). No evidence of hemolysis was ever recorded during the study period in any patient.
Overall AEs were comparable in nature and frequency with other IVIG clinical trials (Berger and Pinciaro 2004). The primary safety objective of the study was to document the incidence of AEs that occurred during or within 1, 24, or 72 h following an infusion of the study product, regardless whether or not the AE was determined to be product related. The primary safety endpoint was analyzed with the intention to demonstrate that the rate of infusions with one or more infusion-related AE is less than 40%. The results demonstrated that the frequency of infusions temporally associated with an AE was 29.2%, which is well below the defined threshold and is comparable with the other IVIG products already in use (Berger and Pinciaro 2004; Berger 2007; Stein et al. 2009; Wasserman et al. 2012; Official Disability Guidelines 2013).
No deaths occurred during the study period, and the 11 serious AEs that occurred in 6 patients during the study period were all clearly assessed as unrelated to the study treatment product.
IVIG-SN was found to be efficacious by meeting the primary endpoint of the study, which is less than 1 serious bacterial infection per patient per year. No acute serious bacterial infections occurred during the entire study period and thus the upper limit of 95% CI was 0.00. Secondary efficacy endpoints were also remarkable for low rates of missed work or school days. The mean rate for the patient missing work or school was 2.6 days per patient per year, which is lower than published reports describing other IVIG products (Berger and Pinciaro 2004; Berger 2007; Stein et al. 2009; Wasserman et al. 2012) and is lower than the average number missed days of work per year by workers in the United States (Official Disability Guidelines 2013; Centers for Disease Control and Prevention 2013). The frequency of visits to a physician's office or emergency room were almost half of the mean of 3.0 predicted in the normal population and were below most published reports in IVIG trials. Very limited use of the therapeutic antibiotics and a low rate of hospitalization were also recorded in this study.
All patients maintained serum IgG trough levels higher than 500 mg/dL with mean concentrations of 925.7 ± 220.2 mg/dL, a level shown to be extremely effective in preventing infections in patients with primary immunodeficiency (Roifman et al. 1987, 2003). Pharmacokinetic studies confirmed good bioavailability of IVIG-SN for total IgG, IgG subclasses, and specific antibodies.
In summary, this clinical trial enrolled patients that were prescreened to confirm the diagnosis of PID and antibody deficiency. Indeed, the trough IgG level prior to any IVIG replacement was 284 ± 218, and all patients (except for patients with agammaglobulinemia) had evidence of inability to respond to vaccinations. The results of IVIG-SN's excellent efficacy and extraordinary tolerability are therefore significant and compare quite favorably to other IVIG products currently available.
REFERENCES
Berger M. Felbogamma® 5% DIF® Investigators. A multicenter, prospective, open label, historically controlled clinical trial to evaluate efficacy and safety in primary immunodeficiency disease patients of Flebogamma 5% DIF®, the next generation of Flebogamma J. Clin. Immunol. 2007 27 6 628 -633
Berger M. and Pinciaro P.H. Flebogamma 5% Investigators. Safety, efficacy, and pharamacokinetics of Flebogamma 5% for replacement therapy in primary immune deficiency J. Clin. Immunol. 2004 24 4 389 -396
Centers for Disease Control and Prevention. 2013. [Online]. FastStats. Available from http://www.cdc.gov/Nchs/fastats/ [accessed 13 January 2014].
Cohn E.J., Strong L.E., Hughes W.L., Mulford D.J., Ashworth J.N., Melin M., and Taylor H.L. Preparation and properties of serum and plasma proteins: a system for the separation into fractions of the protein and lipoprotein components of biological tissues and fluids J. Am. Chem. Soc. 1946 68 3 459 -475
Eibl M.M. History of immunoglobulin replacement Immunol. Allergy Clin. North America. 2008 28 4 737 -764 Editors: Chaim M Roifman, Rafeul Alam
Geha R. and Notarangelo L. The International Union of Immunological Societies (IUIS) Primary Immunodeficiency Disease (PID) Classification Committee J. Allergy Clin. Immunol. 2007 120 4 776 -794
Guidelines for IVIG replacement. 2014. Canadian Immunodeficiency Society. [Online]. Available from http://immunodeficiency.ca/.
Hooper J. Intravenous immunoglobulin evolution of commercial IVIG preparations Immunol. Allergy Clin. North America. 2008 28 765 -768 Editors: Chaim M. Roifman, Rafeul Alam
Melin J.L., Oncley M., and Richert D.A. The separation of the antibodies, isoagglutinins, prothrombin, plasminogen and beta-lipoprotein into subfractions of human plasma J. Am. Chem. Soc. 1949 71 2 541 -550
Official Disability Guidelines. 2013. [Online]. Available from http://www.disabilitydurations.com/pr_repmdc.html [accessed 13 January 2014].
Organ J.S., Grossman W.J., and Navickis R.J. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: A meta-analysis of clinical studies Clin. Immunol. 2010 137 1 21 -30
Roifman C.M. and Gelfand E.W. Replacement therapy with high dose intravenous gammaglobulin improves chronic sinopulmonary disease in patients with hypogammaglobulinemia Pediat. Inf. Dis. J. 1988 7 S92 -S96
Roifman C.M., Levison H., and Gelfand E.W. High dose verses low-dose intravenous immunoglobulin in hypogammaglobinemia and chronic lung disease Lancet. 1987 1 8541 1075 -1077
Roifman C.M., Schroeder H., Berger M., Sorensen R., Ballow M., Buckley R.H., Gewurz A., Korenblat P., Sussman G., and Lemm G. Comparison of the efficacy of IGIV-C, 10% (caprylate/chromatography) and IGIV –SD, 10% as replacement therapy in primary immune deficiency. A randomized double-blind trial Int. Immunopharmacol. 2003 3 9 1325 -1333
Stein M.R., Nelson R.P., Church J.A., Wasserman R.L., Borte M., Vermylen C., and Bichler J. Safety and efficacy of Privigen®, a novel 10% liquid immunoglobulin preparation for intravenous use, in patients with primary immunodeficiency J. Clin. Immunol. 2009 29 1 137 -144
US FDA. 2012. Guidance for Industry: Safety, Efficacy, and Pharmacokinetic Studies to Support Marketing of Immune Globulin Intravenous (Human) as Replacement Therapy for Primary Humoral Immuno-deficiency. [Online]. Available from http://www.fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/blood/ucm072130.htm.
Wasserman R., Church J., and Stein M. Safety, efficacy, and pharmacokinetics of new liquid intravenous immunoglobulin (IVIG) in patients with primary immunodeficiency J. Clin. Immunol. 2012 32 4 663 -669
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 1 • March 2015
Pages: 21 - 29
History
Received: 8 August 2014
Accepted: 20 August 2014
Accepted manuscript online: 29 October 2014
Version of record online: 29 October 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Mark R. Stein, Richard L. Wasserman, James Moy, William Lumry, Eyal Grunebaum, Agnes Nemet, Chaim M. Roifman, and Gordon Sussman. 2015. Efficacy, safety, and tolerability of IVIG-SN in patients with primary immunodeficiency. LymphoSign Journal.
2(1): 21-29. https://doi.org/10.14785/lpsn-2014-0009
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Get Access
Login options
Check if you access through your login credentials or your institution to get full access on this article.
