Novel mutation in PIK3CD affecting the Ras-binding domain
Abstract
Introduction: The phosphoinositide 3-kinase (PI3K) pathway plays critical roles in diverse cellular processes, including differentiation, proliferation, motility, survival, and growth. PI3Kδ, comprised of the catalytic subunit p110δ and regulatory subunit p85α, is essential for normal lymphocyte and myeloid development and function. Gain-of-function mutations in PIK3CD (encoding p110δ) cause a combined immunodeficiency known as activated PI3Kδ syndrome (APDS), in which patients frequently present with recurrent respiratory infections, severe recurrent (or persistent) infections with herpes family viruses, and lymphadenopathy.
Aim: To describe the clinical presentation, immune evaluation, and genetic work-up of 2 patients (daughter and mother) with recurrent sinopulmonary, soft tissue, and skin infections.
Results: Both daughter and mother presented with recurrent sinopulmonary and soft tissue infections. Immune evaluation of the daughter revealed intermittent hypogammaglobulinemia and abnormal specific vaccine responses, while immune parameters of her mother were normal. Whole exome sequencing identified a novel mutation in PIK3CD (NM_005026), c.C719T, resulting in p.T240M. Western blot analysis of downstream AKT levels revealed increased basal phosphorylation, in line with gain-of-function mutations of PIK3CD.
Conclusion: The novel missense mutation in PIK3CD occurs in the region encoding the Ras-binding domain (RBD) of p110δ, and likely alters the structural configuration of the domain. To date, pathogenic mutations targeting the RBD of p110δ have not yet been described. Our results expand on the genotypic spectrum of APDS.
Statement of Novelty: We describe a novel mutation in the Ras-binding domain of PIK3CD leading to a presentation of recurrent sinopulmonary and soft tissue infections in the context of APDS.
Introduction
Phosphoinositide 3-kinase (PI3K) activity is required for normal immune cell development and function, and plays essential roles in diverse cellular processes such as differentiation, proliferation, motility, growth, and survival (Okkenhaug and Vanhaesebroeck 2003; Fruman et al. 2017). The class I PI3K lipid kinases are heterodimers comprising a catalytic subunit (p110α, p110β, or p110δ) and a regulatory subunit (p85α, p55α, p50α, or p55γ), and with the exception of p110δ, are distributed broadly throughout tissues. Under resting conditions, the regulatory subunit stabilizes the catalytic subunit to inhibit downstream signaling activity.
The p110δ subunit, encoded by PIK3CD, is expressed by majority in lymphocytes and myeloid cells (Okkenhaug and Vanhaesebroeck 2003; Fruman et al. 2017). Together with p85α, this heterodimer (termed PI3Kδ) is critical for immune function, including B cell receptor (BCR) and T cell receptor (TCR) signaling (Okkenhaug 2013). Within immune cells, antigen-, cytokine-, costimulatory-, and growth factor receptor-dependent engagement and activation of tyrosine kinases recruits the p85α subunit of PI3Kδ to the cell membrane, thus releasing the inhibitory interaction with p110δ to form active PI3Kδ. PI3Kδ catalyzes the phosphorylation of phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphoinositide-3,4,5-trisphosphate (PIP3), which acts as a membrane tether for signaling proteins with pleckstrin homology (PH) domains. Downstream PI3Kδ signaling targets include BTK (which promotes phospholipase C-dependent responses), PDK1, and AKT (which activates mTOR complex 1 to promote protein synthesis, T cell activation, and differentiation of T cell effector phenotypes, as well as regulating FOXO1-dependent migration of T cells from lymph nodes to the circulation). Dephosphorylation of PIP3 by phosphatase and tensin homolog (PTEN) or inositol 5′-phosphatase terminates the PI3Kδ signal.
Defects in components of the PI3Kδ signaling pathway cause significant functional deficits leading to immunodeficiency and immune dysregulation (Angulo et al. 2013; Lucas et al. 2014). Gain-of-function mutations in PIK3CD and PIK3R1 (encoding the p85α subunit) that were first identified by next generation sequencing techniques cause a combined immunodeficiency known as activated PI3Kδ syndrome (APDS, class 1 or class 2, respectively; OMIM #615513) (Angulo et al. 2013; Lucas et al. 2014, 2016; Crank et al 2014; Takeda et al. 2017; Heurtier et al. 2017; Rae et al. 2017; Wentink 2017; Dulau Florea et al. 2017). APDS is characterized by a wide spectrum of clinical manifestations, although most affected patients present with recurrent respiratory infections and associated lung damage, severe recurrent (or persistent) infections with herpes family viruses, and lymphadenopathy (Elgizouli et al. 2016; Elkaim et al. 2016; Coulter et al. 2017). Individuals with APDS frequently exhibit B and T cell dysfunction, autoimmunity, and are at increased risk of B cell lymphoma (Coulter et al. 2017; Kracker et al. 2014); however, the outcome for these patients vary widely from early death during childhood to adults who remain asymptomatic. Interestingly, rare loss-of-function mutations also present with immunodeficiency (Conley et al. 2012; Zhang et al. 2013).
Here, we describe 2 patients (a daughter and mother) with recurrent sinopulmonary and soft tissue infections in whom a novel missense mutation in the Ras-binding domain (RBD) of PIK3CD was identified.
Methods
Patients
Patient data were compiled prospectively and retrospectively from medical records and entered into the Canadian Centre for Primary Immunodeficiency Registry and Tissue Bank, which has been approved by the SickKids Research Ethics board (protocol No. 1000005598). Consent and assent from each patient and parents were obtained for testing.
Lymphocyte proliferation
Lymphocyte proliferative responses to phytohemagglutinin (PHA) were evaluated. All assays were performed in triplicate and were compared with simultaneously stimulated normal controls, as previously described Sharfe et al. (2014).
Western blotting
Studies were performed on Ficoll-separated peripheral blood lymphocytes. Both patient and control cells were obtained, and either left unstimulated or were stimulated with anti-CD3 antibody (5 μg for 4 × 106 cells) for 10 minutes at 37 °C. Cells were subsequently lysed in 1% Triton X-100 vanadate lysis buffer and protein expression assessed by Western blotting. All blots were repeated at least twice. The primary antibodies used for Western blotting were: Anti-pAKT Ser473 (Invitrogen) and anti-GAPDH (Cell Signaling), followed by appropriate horseradish peroxidase-conjugated secondary antibody. Immunoreactive proteins were visualized by enhanced chemiluminescence.
Whole exome sequencing and variant calling
DNA from blood was submitted to The Centre for Applied Genomics (TCAG), Toronto, Canada, for exome library preparation and sequencing. DNA was quantified by Qubit DNA HS assay (Life Technologies, Carlsbad, CA) and 100 ng of input DNA was used for library preparation using the Ion AmpliSeq Exome Kit (Life Technologies) according to the manufacturer’s recommendations. The Ampliseq Exome library was immobilized on Ion PI™ Ion Sphere™ particles using the Ion PI Template OT2 200 Kit v3. Sequencing was performed with the Ion PI Sequencing 200 Kit v3 and Ion PI Chip v2 in the Ion Proton™ semiconductor sequencing system following the manufacturer’s recommendation.
Alignment and variant calling were performed using Torrent Suite (v4.0) on the Ion Proton Server, using the Ion Proton ampliseq germline low stringency setting and the hg19 reference genome. The variants were annotated using an in-house annotation pipeline based on Annovar (November 2014 version) (Wang et al. 2010) and RefSeq gene models (downloaded from UCSC 01 August 2015).
Sanger sequencing
Patient genomic DNA was extracted from peripheral blood lymphocytes using the Geneaid Genomic DNA Mini Kit. Genomic DNA was amplified by PCR with specific primers designed upstream and downstream of the PI3KCD gene. Sequencing was done using GenomeLab Dye Terminator Cycle Sequencing (DTCS) Quick Start Kit (Beckman Coulter) and analyzed on CEQ 8000 Genetic Analysis System (Beckman Coulter).
Results
Patient 1
The proband presented to medical attention at the age of 4 years. At the time, she presented with recurrent sinopulmonary infections (most notably acute otitis media and sinusitis), and 3 episodes of dental abscesses secondary to caries. She later developed a peri-orbital abscess following traumatic injury, as well as skin infections caused by atypical bacteria (Vagococcus fluvialis and Acinebacter lwoffii). Additionally, she suffered from Haemophilus Influenzae vulvovaginitis requiring treatment with IV antibiotics. Past medical history for this patient included an unremarkable pregnancy with normal prenatal follow-up, term vaginal delivery, a normal neonatal course, and no other medical history. She had been on no medications. Her family history was notable for a mother with recurrent infections (see “Patient 2” below), and a healthy father and brother. There is a history of maternal grandfather with Crohn’s disease, and a paternal grandfather passed away from gastric cancer. There is no history of any other immune disorders in the family.
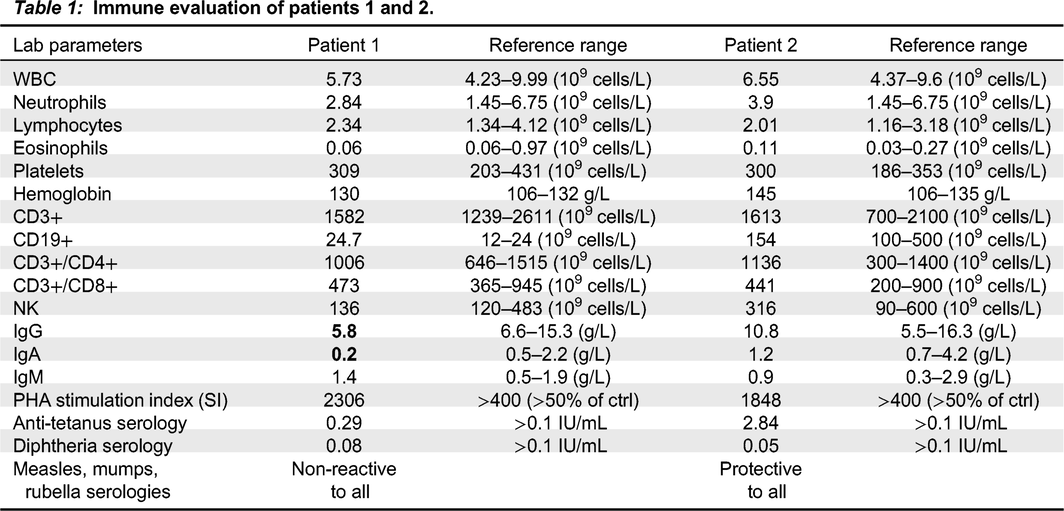
Physical examination demonstrated a well-grown and developing child, with no dysmorphic features, hair, nail or skin abnormalities beyond the reported skin infections, normal tonsillar tissues and lymph nodes, a normal cardiorespiratory exam and no organomegaly. Immune evaluation (Table 1) revealed intermittent hypogammaglobulinemia and abnormal specific vaccine responses, although she has not required immunoglobulin supplementation given spontaneous resolution of sinopulmonary infections with age.
Table 1:
Patient 2
Patient 2, the proband’s mother, is a 42-year-old female with a long-standing history of sinopulmonary infections starting in her early 20s. She suffered frequent sinus and ear infections requiring multiple courses of antibiotics, an episode of facial abscess/cellulitis which progressed to bacteremia, and an episode of dental abscess. She had not suffered from any skin infections as identified in her daughter. She had no other infectious, autoimmune or inflammatory history. As noted above, she had a father with Crohn’s disease but no other notable family history. Her immune evaluation has been unremarkable (Table 1), with normal immunoglobulin levels and vaccine responses.
Genetics
Genetic testing was done via a Primary Immunodeficiency Panel Plus (Blueprint Genetics), followed by research whole exome sequencing. Testing of the proband identified a heterozygous missense variant in PIK3CD (NM_005026), c.C719T, resulting in p.T240M. The substitution of amino acid residue threonine (T) to methionine (M) at position 240 is predicted in-silico to be deleterious or borderline deleterious, resulting in loss of polarity and a longer side chain, and likely altering the structural configuration of the RBD. Targeted sequencing of PIK3CD revealed the identical missense variant in her mother but not her father or brother.
Cell signaling
Peripheral blood lymphocytes (PBL) from patient 2 and a healthy control were stimulated with anti-CD3 antibodies. Western blot analysis showed an increase in basal phosphorylated AKT (pAKT) levels in the patient, consistent with APDS (Figure 1). Levels of pAKT following cell stimulation did not increase further following stimulation, compared with a typical increase in the control sample (Figure 1).
Figure 1:

Discussion
We describe here a mother and daughter with a novel mutation in the RBD of PIK3CD. Their clinical manifestations were typical of APDS, including recurrent sinopulmonary infections, soft tissue and dental abscesses, cellulitis, and humoral deficiency (in the proband) (Coulter et al. 2017).
APDS is caused by gain-of-function mutations in PIK3CD, coding for the p110δ subunit, which is comprised of an adaptor-binding domain (ABD) responsible for binding p85α, a Ras-binding domain (RBD), a C2 domain, a helical domain, and a lipid kinase domain (N-lobe and C-lobe) (Figure 2). Previously reported mutations affected the C2, helical and lipid kinase domains of p110δ, which are the inhibitory contact points for the p85α regulatory subunit (Lucas et al. 2016; Coulter et al. 2017), as well as the ABD and ABD-RBD linker region, responsible for proper ABD orientation (Takeda et al. 2017). These variants lead to overactivation of PI3Kδ, by preventing binding of p110δ with p85α or enhancing the mobilization of p110δ to the cell membrane and increasing the catalytic activity.
Figure 2:

To our knowledge, this is the first variant in the RBD leading to APDS. Our functional assessment has determined this variant to result in a gain-of-function phenotype, as basal AKT phosphorylation was elevated Mandola et al. (2020), with essentially no further phosphorylation noted following stimulation, as frequently seen in gain-of-function mutations affecting other signaling molecules (Sharfe et al. 2015). It is possible that the mutated protein results in enhanced or prolonged binding of Ras-superfamily GTPases to the RBD; however, the precise mechanism leading to this gain-of-function remains to be elucidated. Overall, this report enhances the genotypic spectrum of APDS and proposes a broader range of gain-of-function mutations in PIK3CD underlying this disease.
REFERENCES
Angulo I., Vadas O., Garçon F., Banham-Hall E., Plagnol V., Leahy T.R., Baxendale H., Coulter T., Curtis J., Wu C., Blake-Palmer K., Perisic O., Deborah S., Maes M., Fiddler C., Juss J., Cilliers D., Markelj G., Chandra A., Farmer G., Kielkowska A., Clark J., Kracker S., Debré M., Picard C., Pellier I., Jabado N., Morris J.A., Barcenas-Morales G., Fischer A., Stephens L., Hawkins P., Barrett J.C., Abinun M., Clatworthy M., Durandy A., Doffinger R., Chilvers E.R., Cant A.J., Kumararatne D., Okkenhaug K., Williams R.L., Condliffe A., and Nejentsev S. 2013. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science, 342: 866–871.
Conley M.E., Dobbs A.K., Quintana A.M., Bosompem A., Wang Y.-D., Coustan-Smith E., Smith A.M., Perez E.E., and Murray P.J. 2012. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85alpha subunit of PI3K. J. Exp. Med. 209: 463–470.
Coulter T.I., Chandra A., Bacon C.M., Babar J., Curtis J., Screaton N., Goodlad J.R., Farmer G., Steele C.L., Leahy T.R., Doffinger R., Baxendale H., Bernatoniene J., Edgar J.D.M., Longhurst H.J., Ehl S., Speckmann C., Grimbacher B., Sediva A., Milota T., Faust S.N., Williams A.P., Hayman G., Kucuk Z.Y., Hague R., French P., Brooker R., Forsyth P., Herriot R., Cancrini C., Palma P., Ariganello P., Conlon N., Feighery C., Gavin P.J., Jones A., Imai K., Ibrahim M.A.A., Markelj G., Abinun M., Rieux-Laucat F., Latour S., Pellier I., Fischer A., Touzot F., Casanova J.-L., Durandy A., Burns S.O., Savic S., Kumararatne D.S., Moshous D., Kracker S., Vanhaesebroeck B., Okkenhaug K., Picard C., Nejentsev S., Condliffe A.M., and Cant A.J. 2017. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: A large patient cohort study. J. Allergy Clin. Immunol. 139: 597–606.e4.
Crank M.C., Grossman J.K., Moir S., Pittaluga S., Buckner C.M., Kardava L., Agharahimi A., Meuwissen H., Stoddard J., Niemela J., Kuehn H., and Rosenzweig S.D. 2014. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J. Clin. Immunol. 34: 272–276.
Dulau Florea A.E., Braylan R.C., Schafernak K.T., Williams K.W., Daub J., Goyal R.K., Puck J.M., Koneti Rao V., Pittaluga S., Holland S.M., Uzel G., and Calvo K.R. 2017. Abnormal B-cell maturation in the bone marrow of patients with germline mutations in PIK3CD. J. Allergy Clin. Immunol. 139: 1032–1035.e6.
Elgizouli M., Lowe D.M., Speckmann C., Schubert D., Hülsdünker J., Eskandarian Z., Dudek A., Schmitt-Graeff A., Wanders J., Jørgensen S.F., Fevang B., Salzer U., Nieters A., Burns S., and Grimbacher B. 2016. Activating PI3Kdelta mutations in a cohort of 669 patients with primary immunodeficiency. Clin. Exp. Immunol. 183: 221–229.
Elkaim E., Neven B., Bruneau J., Mitsui-Sekinaka K., Stanislas A., Heurtier L., Lucas C.L., Matthews H., Deau M.-C., Sharapova S., Curtis J., Reichenbach J., Glastre C., Parry D.A., Arumugakani G., McDermott E., Kilic S.S., Yamashita M., Moshous D., Lamrini H., Otremba B., Gennery A., Coulter T., Quinti I., Stephan J.-L., Lougaris V., Brodszki N., Barlogis V., Asano T., Galicier L., Boutboul D., Nonoyama S., Cant A., Imai K., Picard C., Nejentsev S., Molina T.J., Lenardo M., Savic S., Cavazzana M., Fischer A., Durandy A., and Kracker S. 2016. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase delta syndrome 2: A cohort study. J. Allergy Clin. Immunol. 138: 210–218.e9.
Fruman D.A., Chiu H., Hopkins B.D., Bagrodia S., Cantley L.C., and Abraham R.T. 2017. The PI3K Pathway in Human Disease. Cell, 170: 605–635.
Heurtier L., Lamrini H., Chentout L., Deau M.-C., Bouafia A., Rosain J., Plaza J.-M., Parisot M., Dumont B., Turpin D., Merlin E., Moshous D., Aladjidi N., Neven B., Picard C., Cavazzana M., Fischer A., Durandy A., Stephan J.-L., and Kracker S. 2017. Mutations in the adaptor-binding domain and associated linker region of p110delta cause Activated PI3K-delta Syndrome 1 (APDS1). Haematologica, 102: e278–e281.
Kracker S., Curtis J., Ibrahim M.A.A., Sediva A., Salisbury J., Campr V., Debré M., Edgar J.D.M., Imai K., Picard C., Casanova J.-L., Fischer A., Nejentsev S., and Durandy A. 2014. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase delta syndrome. J. Allergy Clin. Immunol. 134: 233–236.e3.
Lucas C.L., Kuehn H.S., Zhao F., Niemela J.E., Deenick E.K., Palendira U., Avery D.T., Moens L., Cannons J.L., Biancalana M., Stoddard J., Ouyang W., Frucht D.M., Koneti Rao V., Atkinson T.P., Agharahimi A., Hussey A.A., Folio L.R., Olivier K.N., Fleisher T.A., Pittaluga S., Holland S.M., Cohen J.I., Oliveira J.B., Tangye S.G., Schwartzberg P.L., Lenardo M.J., and Uzel G. 2014. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat. Immunol. 15: 88–97.
Lucas C.L., Chandra A., Nejentsev S., Condliffe A.M., and Okkenhaug K. 2016. PI3Kdelta and primary immunodeficiencies. Nat. Rev. Immunol. 16: 702–714.
Mandola A.B., Dadi H., Reid B., and Roifman C.M. 2020. Novel heterozygous PIK3CD mutation presenting with only laboratory markers of combined immunodeficiency. LymphoSign J. 7: 49–55.
Okkenhaug K. 2013. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol. 31: 675–704.
Okkenhaug K. and Vanhaesebroeck B. 2003. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 3: 317–330.
Rae W., Gao Y., Ward D., Mattocks C.J., Eren E., and Williams A.P. 2017. A novel germline gain-of-function variant in PIK3CD. Clin. Immunol. 181: 29–31.
Sharfe N., Nahum A., Newell A., Dadi H., Ngan B., Pereira S.L., Herbrick J.-A., and Roifman C.M. 2014. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1. J. Allergy Clin. Immunol. 133: 807–817.
Sharfe N., Merico D., Karanxha A., Macdonald C., Dadi H., Ngan B., Herbrick J.-A., and Roifman C.M. 2015. The effects of RelB deficiency on lymphocyte development and function. J. Autoimmun. 65: 90–100.
Takeda A.J., Zhang Y., Dornan G.L., Siempelkamp B.D., Jenkins M.L., Matthews H.F., McElwee J.J., Bi W., Seeborg F.O., Su H.C., Burke J.E., and Lucas C.L. 2017. Novel PIK3CD mutations affecting N-terminal residues of p110delta cause activated PI3Kdelta syndrome (APDS) in humans. J. Allergy Clin. Immunol. 140: 1152–1156.e10.
Wang K., Li M., and Hakonarson H. 2010. Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 11: 843–854.
Wentink M., Dalm V., Lankester A.C., van Schouwenburg P.A., Schölvinck L., Kalina T., Zachova R., Sediva A., Lambeck A., Pico-Knijnenburg I., van Dongen J.J.M., Pac M., Bernatowska E., van Hagen M., Driessen G., and van der Burg M. 2017. Genetic defects in PI3Kdelta affect B-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin. Immunol. 176: 77–86.
Zhang K, Husami A, Marsh R, and Jordan MB. 2013. Identification of a phosphoinositide 3-kinase (PI-3K) p110δ (PIK3CD) deficient individual. J. Clin. Immunol. 33: 673–674.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 9 • Number 1 • March 2022
Pages: 11 - 16
History
Received: 14 February 2022
Accepted: 22 February 2022
Accepted manuscript online: 23 February 2022
Copyright
© 2022.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
LauraAbrego Fuentes, JennyGarkaby, OriScott, JessicaWillet Pachul, HarjitDadi, and LindaVong. 2022. Novel mutation in PIK3CD affecting the Ras-binding domain. LymphoSign Journal.
9(1): 11-16. https://doi.org/10.14785/lymphosign-2022-0002
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Get Access
Login options
Check if you access through your login credentials or your institution to get full access on this article.
