A novel mutation in TRAC in a patient with abnormal newborn screening for severe combined immunodeficiency
Abstract
Background: The T cell receptor (TCR)-α chain plays a key role in TCR structure and function. Biallelic mutations in TRAC, encoding the constant region of the TCR-α chain, obliterates TCR expression and results in immunodeficiency. TCR-α chain deficiency presents at infancy or childhood with repeated viral and bacterial infections, enlarged liver, spleen, and lymph nodes as well as autoimmune features and lymphoma (OMIM #615387).
Aim: To broaden the genotypic and phenotypic spectrum of TCR-α chain deficiency.
Methods: We present a case report of a patient with severe combined immunodeficiency (SCID) due to a novel autosomal recessive mutation in TRAC.
Results: Our patient was identified at 13 days of life due to abnormal T cell receptor excision circle levels detected by newborn screening (NBS). Immune evaluation revealed profound lymphopenia, depressed responses to the mitogen PHA and a skewed T cell repertoire, all consistent with SCID. The patient was found to carry a novel homozygous mutation in the TRAC gene.
Conclusion: A novel homozygous mutation in the TRAC gene caused profound T cell lymphopenia and aberrant in vitro mitogenic response, the hallmarks of SCID.
Statement of Novelty: TCR-α chain deficiency is a rare and relatively new condition and not very well defined. We herein report a novel mutation in TRAC resulting in SCID.
Introduction
Several genetic defects lead to the clinical syndrome of severe combined immunodeficiency (SCID), characterized by profound susceptibility to bacterial, viral, and opportunistic infections, failure to thrive, and death in infancy in the absence of appropriate treatment. SCID is caused by genetic defects that affect the T cell receptor (TCR), T cell differentiation, maturation, survival, or function (Roifman 2019). The TCR is composed of two subunits with discrete functions: (1) an antigen-recognizing heterodimer consisting either α and β chains or γ and δ chains (each with a variable region and constant region), and (2) a signal transduction unit composed of three invariant CD3 dimers (Gouaillard et al. 2001). The TRAC gene encodes for the constant region of the TCR-α chain. The first cases of TCR-α chain deficiency were reported by Morgan et al. (2011) in 2 unrelated children from consanguineous families of Pakistani descent. They presented in infancy with recurrent respiratory infections, otitis media, candidiasis, diarrhea, and failure to thrive. Both patients also had features of immune dysregulation and were treated with hematopoietic stem cell transplantation (HSCT) (Morgan et al. 2011). In a recent case series, Rawat et al. (2021) reported a nonconsanguineous East Indian family in which 3 siblings had an immunodeficiency disorder resulting in death in all patients aged between 12 months and 11 years. Five patients have been so far identified with TCR-α chain deficiency. All carry an identical homozygous G to A transition in exon 3, located in the consensus 5’ splice site, predicting aberrant transcription, skipping of exon 3, and a protein that is lacking significant portions of the transmembrane and cytoplasmic domains of the TCR-α chain.
TCR-α chain deficiency is a rare and relatively new condition that remains ill-defined, and is attributed to mutations in the TRAC gene. We herein report a novel mutation in TRAC resulting in SCID.
Case presentation
Clinical case
An asymptomatic newborn male was found to have low T cell receptor excision circles (TREC) by the Ontario newborn screening (NBS) program for SCID.
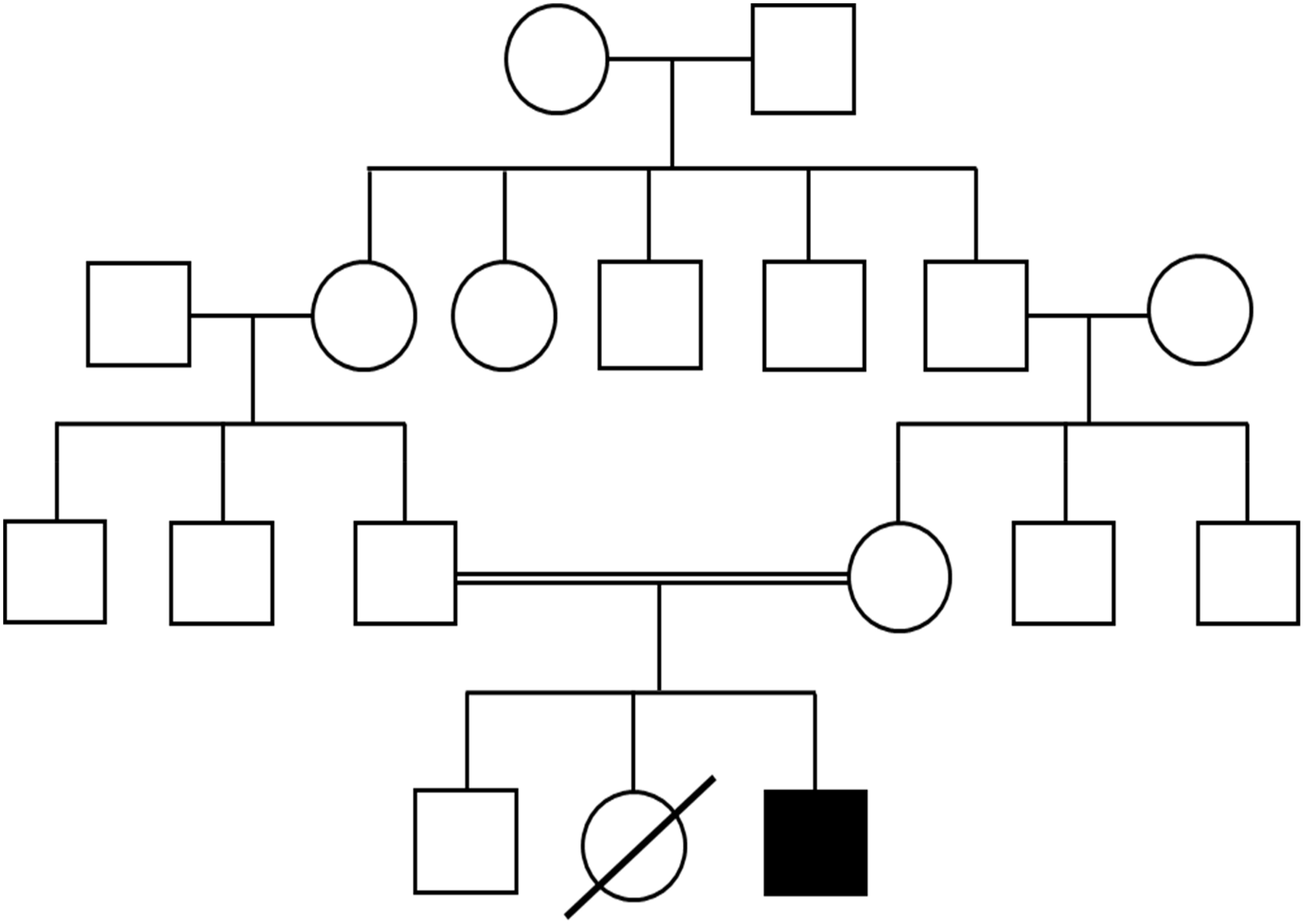
The infant was the product of consanguineous parents of East Indian descent. He was born at 35 + 6 weeks after induced vaginal delivery due to concern for intrauterine growth restriction and had an uneventful postnatal course. His birth weight was 2060 gr (<3rd WHO percentile). The family history was remarkable for a female sibling who died in India at 8 months of age with a history of recurrent bacterial infections, oral thrush, diarrhea, and failure to thrive. His parents were healthy (Figure 1). On physical exam the patient had no palpable lymph nodes and lacked dysmorphic features. His growth and development appeared normal.
Figure 1:
Investigations
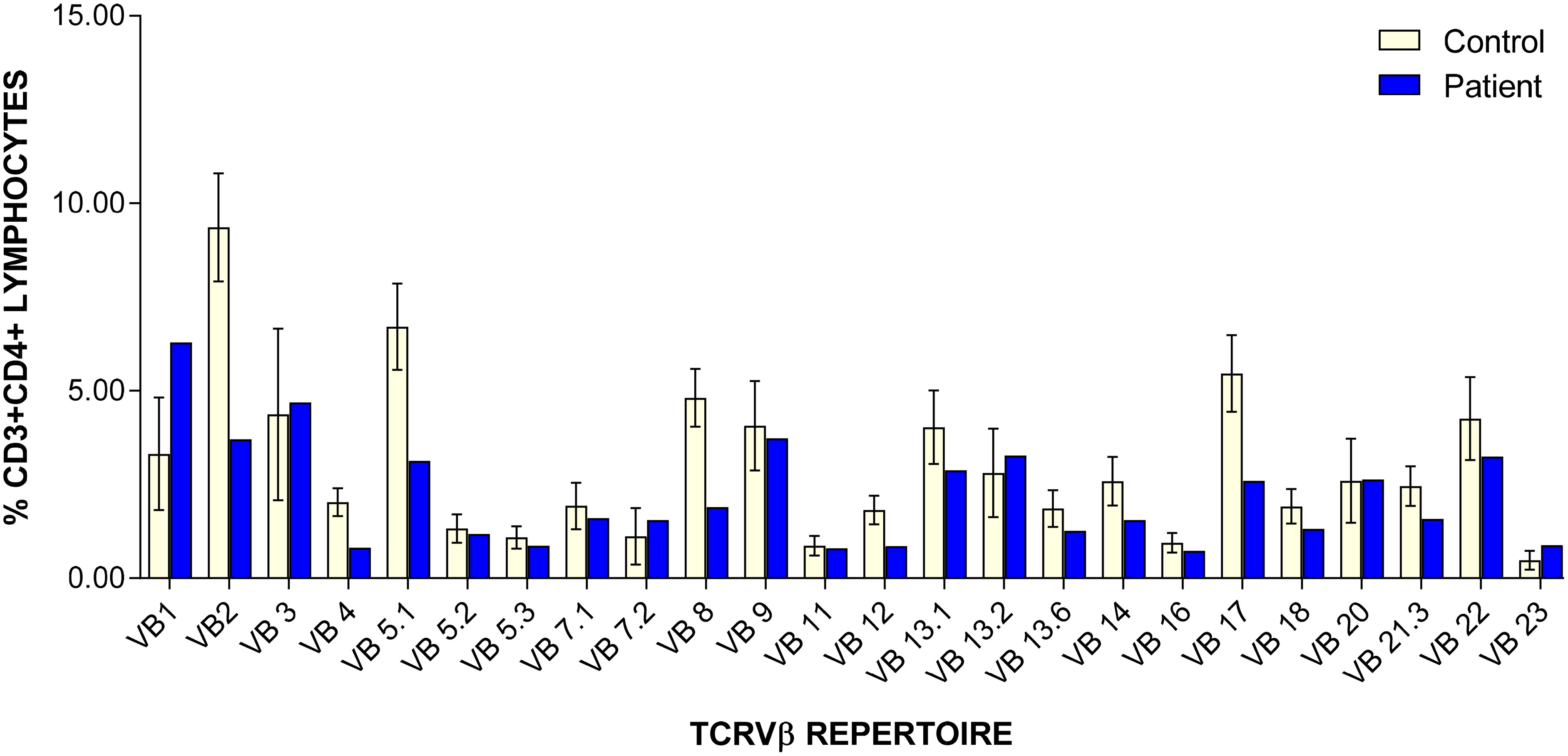
TREC levels were undetectable (0 copies; cut-off >75). Further analysis of adenosine deaminase (ADA) and purine nucleoside phosphorylase (PNP) metabolite profiles were normal, and targeted testing for mutations in IKBKB and Zap70 were negative. Complete blood count and differential were normal apart from mildly low lymphocytes count of 1.79 × 109/L (normal: 1.96–8.94 × 109/L). Lymphocyte immunophenotyping was abnormal, with extremely low CD3+CD4+ (113 cells/μL; normal: 1700–5300 cells/μL) and CD3+CD8+ (76 cells/μL; normal: 400–1700 cells/μL) counts. CD19+ cells were normal (638 cells/μL; normal: 600–1900 cells/μL), as were CD16+CD56+ cells (710 cells/μL; normal: 186–724 cells/μL). PHA stimulation index was severely depressed at 33 (control: 2010), T cell receptor Vβ (TCRVβ) repertoire showed under representation of most CD4+ Vβ families and absent representation of all the CD8+ Vβ families (Figures 2 and 3). Analysis of naïve/memory T cells (CD45RA/RO) was abnormal with predominance of memory cells and low percentage of naïve cells. Karyotyping was negative for maternal engraftment.
Figure 2:

Figure 3:
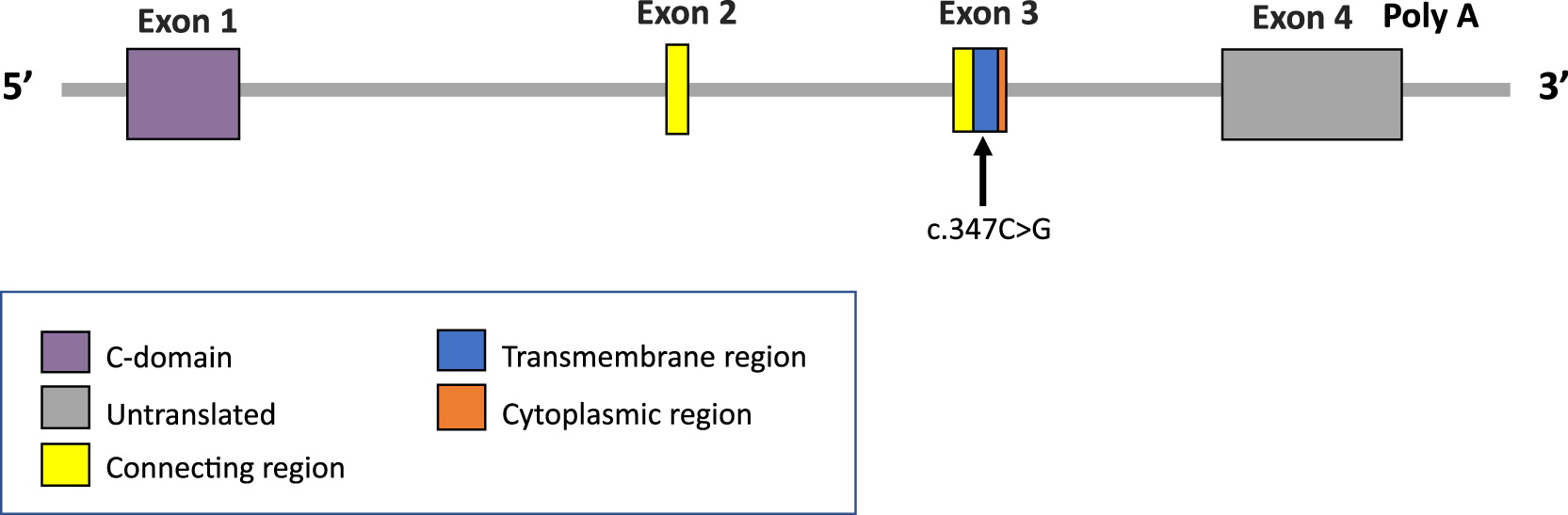
Genetic analysis began with normal karyotype and normal in-situ hybridization for 22q11.2. Further analysis involved a primary immunodeficiency panel, which revealed a nonsense homozygous variant in exon 3 of the TRAC gene, c.C347G; p.Ser116Ter (Figure 4). This variant was identified in 2 of 234 700 alleles from presumed healthy individuals in large population databases (GnomAD allele frequency 0.00085%) and has not been reported in the literature or in ClinVar. The variant results in the introduction of a premature termination codon, predicted to cause a truncation of the encoded protein or absence of the protein due to nonsense-mediated decay which are known mechanisms of disease (Shelton et al. 2001).
Figure 4:
An additional finding on the panel was a missense variant detected in the RAC2 gene, c.C545T; p.Thr182Met. This variant has not been reported in affected individuals in the literature. In ClinVar, one laboratory considers this a variant of unknown significance (VUS) and another classifies it as likely benign. This variant was identified in 32 of 281 160 alleles from presumed healthy controls in large population databases (GnomAD allele frequency 0.011%). Mutations in this gene are associated with neutrophil immunodeficiency syndrome (OMIM# 608203, 618987, 618986), which clearly does not match the features observed in this patient, and was therefore considered irrelevant.
Outcome
IVIG replacement therapy and PJP prophylaxis were commenced, and HSCT offered.
Discussion
The development and implementation of NBS have led to increased early detection of patients with SCID, allowing affected infants to be treated appropriately with HSCT before the onset of complications. As reported previously, X-linked IL2RG SCID is most frequently observed, while other causes of SCID include JAK3 mutations, ADA deficiency, deficiency of RAG1 or RAG2, IL7R deficiency, TCR CD3 δ, ε, and ξ chains deficiencies, Artemis mutations, and syndromic SCID such as Cartilage hair hypoplasia. Despite advanced genetic testing of known SCID genes, up to 10% of infants with SCID remain without a proven genotype. In this regard, homozygous mutations in TRAC resulting in TCR-α chain constant deficiency were not previously reported in the context of positive NBS (Kwan et al. 2014; Dinur et al. 2019; Mandola et al. 2019; Giżewska et al. 2020; Kumrah et al. 2020; Scott et al. 2021).
The first report of mutations in TRAC to cause immunodeficiency was by Morgan et al. in 2011 describing 2 patients. Both patients displayed features of combined immunodeficiency with recurrent bacterial infections, candidiasis, diarrhea, and failure to thrive. One patient also displayed chronic EBV, varicella, and human herpesvirus 6 (HHV6) viremia while the other patient was able to clear varicella at the age of 6 years uneventfully. In addition to immunodeficiency, both patients had evidence of immune dysregulation with hypereosinophilia, low-titer antinuclear antibodies (ANA), vitiligo, and alopecia areata in one patient and hypereosinophilia, eczema, autoimmune hemolytic anemia, anti-TTG antibodies, low-titer ANA in the other patient. Interestingly, humoral immunity against vaccine antigens appeared normal despite T cells abnormalities of abnormal CD3+ cells which expressed TCRαβ at extremely low levels and low PHA stimulation index (<50% of the control). When assessed by flow cytometry, surface staining for TCR-αβ was strongly reduced in patient cells. Functional studies showed that the TRAC mutation resulted in mis-localization of the TCR-α and -β chains. Furthermore, patient cells showed undetectably low expression of both TCR-α or TCR-β polypeptides, suggesting that the TRAC mutation not only resulted in TCR-α deficiency but also reduced TCR-β expression.
An additional recent case series of 3 patients was reported by Rawat et al. in 2021. The patients were 3 siblings born to a non-consanguineous family from Northwest India who all succumbed due to complications of immunodeficiency. All three had similar clinical and immunological features, manifesting as recurrent ear and chest infections, deep seated abscesses, cutaneous warts and lymphadenopathy, however, the youngest child also developed non-Hodgkin’s lymphoma in infancy. Genetic analysis revealed a previously reported variant in the TRAC gene (Rawat et al. 2021).
Clinically, our patient displayed lymphopenia and PHA < 10% of the control, in complete agreement with a diagnosis of T-B+NK+ SCID (Shearer et al. 2015), and has been well for the first months of life with supportive care until he received a HSCT. This patient would likely have presented with recurrent life-threatening infections as described thus far in the literature with low overall survival rate had it not been for the early detection through NBS.
The β chain gene is rearranged before the α chain, and, if productive, the TCR-β chain is initially expressed with pre-T-α, the invariant chain. Functional signaling through this complex must occur for progression of α chain rearrangement to take place (Anderson et al. 1996). Furthermore, defective expression of TCR-α would be expected to impair αβ T cell development beyond β selection because of a failure to deliver positively selecting signals as was shown by Morgan et al. (2011).
Our patient’s variant, a nonsense homozygous variant in exon 3 of the TRAC gene, c.C347G; p.Ser116Ter, results in a premature termination codon in exon 3, predicted to cause truncation of the encoded protein or complete absence. As the TRAC gene encodes for the constant region of the TCR-α chain and is essential for membrane expression of the TCR-αβ heterodimer, we presume that in our patient this probably resulted in a non-functional TCR-α chain, which is essential for T cell development, explaining the T-B+NK+ phenotype as well as the near complete lack of representation of CD4+ and CD8+ Vβ families, respectively.
The current report contributes to the spectrum of mutations in TRAC, both genotypically and phenotypically. Furthermore, to our knowledge, this is the first of case of a mutation in TRAC reported in regard to positive NBS for SCID.
REFERENCES
Anderson G., Moore N.C., Owen J.J.T., and Jenkinson E.J. 1996. Cellular interactions in thymocyte development. Annu. Rev. Immunol. 14: 73–99.
Dinur Schejter Y., Mandola A., and Reid B. 2019. Coronin 1A deficiency identified by newborn screening for severe combined immunodeficiency. LymphoSign J. 6(1): 17–25.
Giżewska M., Durda K., Winter T., Ostrowska I., Ołtarzewski M., Klein J., Blankenstein O., Romanowska H., Krzywińska-Zdeb E., Patalan M.F., Bartkowiak E., Szczerba N., Seiberling S., Birkenfeld B., Nauck M., von Bernuth H., Meisel C., Bernatowska E.A., Walczak M., and Pac M. 2020. Newborn screening for SCID and other severe primary immunodeficiency in the Polish-German Transborder area: Experience from the first 14 months of collaboration. Front. Immunol. 11(October).
Gouaillard C., Huchenq-Champagne A., Arnaud J., Chen C.-H., and Rubin B. 2001. Evolution of T cell receptor (TCR) αβ heterodimer assembly with the CD3 complex. Eur. J. Immunol. 31(12): 3798–3805.
Kumrah R., Vignesh P., Patra P., Singh A., Anjani G., Saini P., Sharma M., Kaur A., and Rawat A. 2020. Genetics of severe combined immunodeficiency. Genes Dis. 7(1): 52–61.
Kwan A., Abraham R.S., Currier R., Brower A., Andruszewski K., Abbott J.K., Baker M., Ballow M., Bartoshesky L.E., Bonagura V.R., Bonilla F.A., Brokopp C., Brooks E., Caggana M., Celestin J., Church J.A., Comeau A.M., Connelly J.A., Cowan M.J., Cunningham-Rundles C., Dasu T., Dave N., De La Morena M.T., Duffner U., Fong C.-T., Forbes L., Freedenberg D., Gelfand E.W., Hale J.E., Hanson I.C., Hay B.N., Hu D., Infante A., Johnson D., Kapoor N., Kay D.M., Kohn D.B., Lee R., Lehman H., Lin Z., Lorey F., Abdel-Mageed A., Manning A., McGhee S., Moore T.B., Naides S.J., Notarangelo L.D., Orange J.S., Pai S.-Y., Porteus M., Rodriguez R., Romberg N., Routes J., Ruehle M., Rubenstein A., Saavedra-Matiz C.A., Scott G., Scott P.M., Secord E., Seroogy C., Shearer W.T., Siegel S., Silvers S.K., Stiehm E.R., Sugerman R.W., Sullivan J.L., Tanksley S., Tierce M.L., Verbsky J., Vogel B., Walker R., Walkovich K., Walter J.E., Wasserman R.L., Watson M.S., Weinberg G.A., Weiner L.B., Wood H., Yates A.B., and Puck J.M. 2014. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. J. Am. Med. Assoc. 312(7): 729–738.
Mandola A.B., Reid B., Sirror R., Brager R., Dent P., Chakroborty P., Bulman D.E., and Roifman C.M. 2019. Ataxia Telangiectasia diagnosed on newborn screening–case cohort of 5 years’ experience. Front. Immunol. 10(December): 1–7.
Morgan N., Goddard S., Cardno T.S., McDonald D., Rahman F., Barge D., Ciupek A., Straatman-Iwanowska A., Pasha S., Guckian M., Anderson G., Huissoon A., Cant A., Tate W.P., Hambleton S., and Maher E.R. 2011. Mutation in the TCR [alpha] subunit constant gene (TRAC) leads to a …. J. Clin. Invest. 121(2): 695–702.
Rawat A., Singh A., Dobbs K., Pala F., Delmonte O.M., Vignesh P., Jindal A.K., Gupta A., Suri D., Kaur A., Shandilya J.K., Sachdeva M.U.S., Walia M., Regueiro J.R., Briones A.C., Notarangelo L.D., and Singh S. 2021. Skewed TCR Alpha, but not Beta, Gene Rearrangements and Lymphoma Associated with a Pathogenic TRAC Variant. J. Clin. Immunol. 41: 1395–1399.
Roifman, C.M. 2019. Primary T-cell immunodeficiencies. Fifth edit, clinical immunology. 5th ed. Elsevier Ltd.
Scott O., Garkaby J., Willett-Pachul J., Mandola A.B., and Pasternak Y. 2021. A novel splice site variant in FOXN1 in a patient with abnormal newborn screening for severe combined immunodeficiency and congenital lymphopenia. LymphoSign J. 8: 1–4.
Shearer W.T., Dunn E., Notarangelo L.D., Dvorak C.C., Puck J.M., Logan B.R., Griffith L.M., Kohn D.B., O’Reilly R.J., Fleisher T.A., Pai S.-Y., Martinez C.A., Buckley R.H., and Cowan M.J. 2015. Scid, and Omenn syndrome : The primary immune. 133(4): 1092–1098.
Shelton J.G., Gülland S., Nicolson K., Kearsea K.P., and Bäckström T. 2001. Importance of the T cell receptor alpha-chain transmembrane distal region for assembly with cognate subunits. Mol. Immunol. 38(4): 259–265.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 9 • Number 1 • March 2022
Pages: 5 - 10
History
Received: 10 February 2022
Accepted: 20 February 2022
Accepted manuscript online: 25 February 2022
Copyright
© 2022.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
JennyGarkaby, Laura EdithAbrego Fuentes, JessicaWillett Pachul, AbbyWatts-Dickens, and MeghanFraser. 2022. A novel mutation in TRAC in a patient with abnormal newborn screening for severe combined immunodeficiency. LymphoSign Journal.
9(1): 5-10. https://doi.org/10.14785/lymphosign-2022-0001
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Get Access
Login options
Check if you access through your login credentials or your institution to get full access on this article.
