Wiskott-Aldrich syndrome caused by a novel mutation in the WAS gene and presenting with a mild phenotype
Abstract
Background: Wiskott-Aldrich syndrome (WAS) is an X-linked recessive disorder associated with combined immunodeficiency, microthrombocytopenia, eczema, and an increased risk of autoimmunity and cancer.
Aim: To report the clinical presentation, immune features, and genetic mutation in a patient with a novel mutation in the Wiskott-Aldrich syndrome (WAS) gene, causing a mild phenotype of WAS.
Methods: The patient’s chart was reviewed. We report the phenotypical and laboratory characteristics of a patient with a mild phenotype of WAS identified by WAS gene sequence analysis.
Results: Our patient presented with thrombocytopenia and 3 episodes of otitis media at 24 months of age, with no other significant manifestations suggestive of immunodeficiency or immune dysregulation. A missense mutation was found in exon 12 of the WAS gene, C1498>T, leading to a Trp500Arg amino acid change. Currently, he is 15 years old and remains in good health, free of infections or other complications to date.
Conclusion: Genetic analysis is helpful for the diagnosis of WAS; our patient’s mutation was found to cause a mild phenotype.
Statement of novelty: We describe a patient with a mild phenotype of WAS with a novel mutation in the WAS gene, thus, expanding the spectrum of WAS gene mutations.
Introduction
Wiskott-Aldrich syndrome (WAS) is a rare X-linked primary immunodeficiency disorder characterized by recurrent infection, eczema, microthrombocytopenia and an increased risk of autoimmune disorders and malignancies, mostly lymphoma Buchbinder et al. 2014. The Wiskott-Aldrich syndrome gene (WAS) is located at Xp11.22–p11.23 and contains 12 exons, encoding the Wiskott-Aldrich syndrome protein (WASp) — a 502-amino acid intracellular multi-domain protein, which has a crucial role in cytoskeleton organization and cell signaling (Massaad et al. 2013; Kirchhausen and Rosen 1996). WASp contains 5 distinct structural domains: the N-terminal WASP-homology domain 1 (WH1), the GTPase-binding domain (GBD), the proline-rich region (PRR), the verprolin homology domain (V), and the cofilin-homology sequence (C), and a C-terminal acidic (A) domain (Kim et al. 2000; Massaad et al. 2013) (Figure 1).
Figure 1:
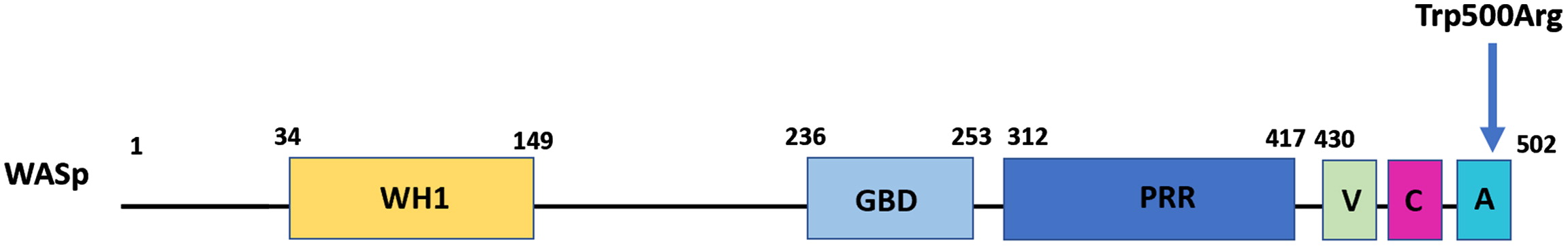
The N-terminal domain of WASp is important for IL-2 signaling while the WH1 domain is the binding site of WASp-interacting protein (WIP). WIP is crucial for the stability of WASp. The inactive state of WASp is maintained by autoinhibition with binding of the VCA domain to the GBD domain. The PRR domain serves as a docking site for other signaling and adaptor proteins. The C-terminal VCA domain is the functional unit that has a critical role in the process of actin polymerization (Kim et al. 2000; Chou et al. 2006). There are multiple immunological functions which are dependent on the normal actin cytoskeleton, including lymphoid cell proliferation, immune synapse assembly and signaling, lymphoid and myeloid cell migration, as well as natural killer cell cytolytic activity and phagocytosis (Rivers et al. 2017).
The clinical manifestations of WAS include microthrombocytopenia associated with increased risk of bleeding, eczema in different degrees of severity, and recurrent infections. Infections may include severe life-threatening viral infections, bacterial, and opportunistic infections (Imai et al. 2004). Furthermore, patients with WAS are at high risk of developing autoimmune complications such as autoimmune hemolytic anemia, arthritis, inflammatory bowel disease, and vasculitis (Dupuis-Girod et al. 2003).
Different mutations in the WAS gene have been identified, resulting in various phenotypes and a broad range of disease severity (Imai et al. 2003). Most of the missense mutations identified in WAS patients are located in exons one to four, affecting the WH1 domain, leading to poor binding of WIP to the WH1 domain and WASp instability (Luthi et al. 2003). Missense mutations usually result in normal-sized WASp, often with reduced protein expression and were considered to cause a clinical phenotype less severe than patients with no WASp expression, with some exceptions. In contrast, patients with nonsense mutations either lack WASp or express a truncated protein, and were considered to present with the classical phenotype of WAS. However, data provided in published reports have so far failed to demonstrate a clear correlation of genotype/protein expression and phenotype (Imai et al. 2003, 2004; Albert et al. 2010).
In some cases, WAS can be fatal without hematopoietic stem cell transplantation early in life, whereas milder cases can be managed symptomatically with clinical follow up and/or immunoglobulin replacement therapy (Imai et al. 2004). In this case, we report on a young male with WAS caused by a novel mutation. He presented with a mild phenotype and has no WAS-related complications.
Methods
Patient chart review and targeted sequencing of the WAS gene were performed following informed consent, in accordance with a research ethics approved protocols. Following our patient’s diagnosis, further genetic testing was performed on the patient’s mother, maternal grandfather, and sibling for the known familial mutation. Targeted sequencing was the preferred method at the time of diagnosis as commercially available genetic panels were not available.
Results
Case presentation
Our patient, currently a 15-year-old male, presented at the age of 2 years with thrombocytopenia and mild bruising. He was initially diagnosed with immune thrombocytopenic purpura but further findings of persistent low platelet count, microthrombocytopenia on blood smear, and 3 episodes of otitis media prompted immunological evaluation. He did not experience any severe or deep-seated infections, nor dermatitis, or diarrhea. He was born to non-consanguineous healthy parents. His family history is significant for a maternal grandfather with Crohn’s disease and frequent epistaxis. His physical exam was within normal limits except for mild bruising. He had normal growth and development.
Investigations
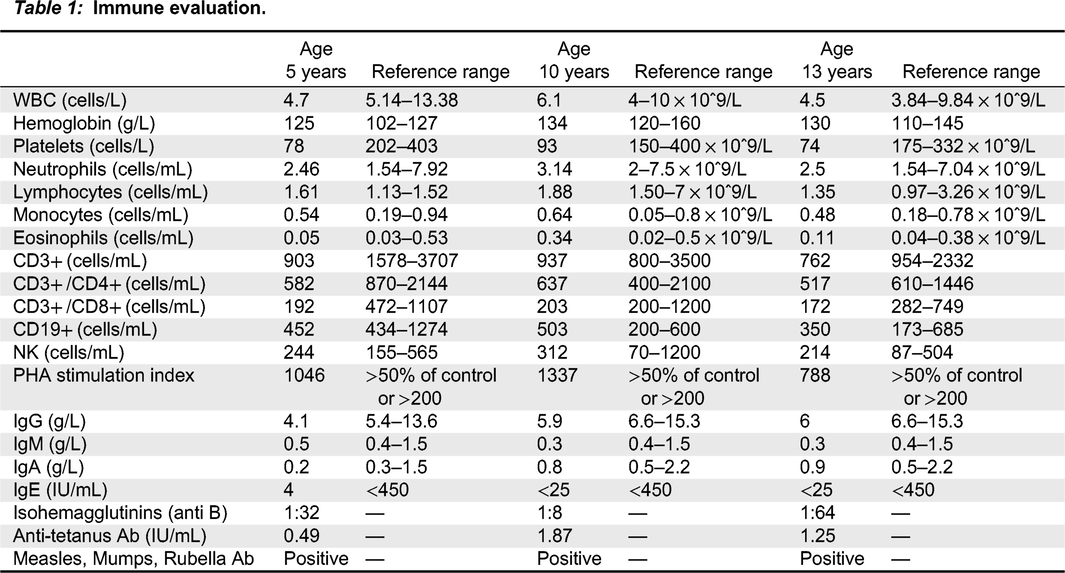
Immune evaluation revealed normal white blood count (WBC), no anemia, and thrombocytopenia of 50 × 109/L. Total lymphocyte and T cell counts were normal (Table 1), but over time, a gradual reduction in both CD4+ and CD8+ cells was observed. PHA stimulation index was slightly low, however, repeated testing was within the normal range. Serum concentrations of immunoglobulins (IgG, IgA, and IgM) were normal for age and vaccine responses to Measles, Mumps, Rubella and Tetanus toxoid were protective. WAS gene sequencing revealed a missense mutation of C1498>T, leading to a Trp500Arg amino acid change. This mutation has not been described in the literature previously. Flow cytometry showed WASp was present, but reduced expression of WASp in leucocytes was noted.
Table 1:
Further genetic testing via sequencing the region covering the known familial mutation was performed on the patient’s maternal grandfather, healthy mother, and healthy sibling. Both the grandfather and sibling were found to be negative, and both had normal platelet counts. The patient’s mother was found to be a carrier of the same mutation. She is otherwise well and healthy.
Outcome
Further evaluation revealed that the patient’s brother was not a match for hematopoietic stem cell transplantation. The challenge of future management for this patient stemmed from the fact that, at the time, his prognosis was uncertain given that his mutation had not been previously described in the literature. Since he did not have a matched-sibling donor, nor a matched-unrelated donor, hematopoietic stem cell transplantation did not become part of his treatment. Over the following 13 years, he continued to be well, with no recurrent infections. He did not develop clinical evidence suggestive of immune dysregulation, i.e., inflammatory bowel disease, arthritis, vasculitis, autoimmune hemolytic anemia etc. His immunological evaluation is significant for stable thrombocytopenia and T cell lymphopenia, borderline immunoglobulins levels and normal vaccines response.
Discussion
WAS is a rare immunodeficiency with estimated prevalence of ∼1:250,000, male births in Canada and the U.S. (Perry et al. 1980). It is characterized by recurrent infections, eczema and microthrombocytopenia, as well as an increased risk for cancer and autoimmunity (Massaad et al. 2013; Rivers et al. 2017). The causative gene, WAS, encodes WASp, which is a hematopoietic cell specific protein and is involved in actin polymerization, cytoskeletal rearrangement, and signaling events (Massaad et al. 2013).
Our patient’s mutation is at the C-terminal acidic A domain which is required for the initiation of actin polymerization and thus normal cell motility. Deletion of the VCA domain results in complete loss of WASp mediated actin polymerization (Massaad et al. 2013). Another important role of the VCA domain is in WASp autoinhibition, which occurs upon GBD site binding to the C-terminal VCA region, resulting in autoinhibition of the stimulatory activity of the protein (Scherl et al. 2002).
The mild phenotype observed in our patient is most probably associated with intact WASp functions, such as the WIP/WH1 domain interaction and protein stabilization, signaling through the GBD domain, and normal docking at the PRR domain, as those are not affected by the far C-terminal acidic mutation. Phosphorylation of WASp on tyrosine 291, a location that is most probably not affected by our patient’s mutation, was shown to enhance the actin polymerization activity of WASp via the Actin Related Protein (Arp) 2/3 complex. Moreover, immune functions such as T cell differentiation, memory B cell activation, and transcription of inflammatory cytokines are independent of the normal actin polymerization by WASp (Rivers et al. 2017).
According to the scoring system suggested by Zhu et al. (1997), patients considered to have an X-linked thrombocytopenia (XLT) phenotype are assigned a score of one to two, whereas patients considered to have WAS are assigned a score of three to four. XLT and WAS patients who develop autoimmunity and/or malignancies at a later stage in life progress to a score of five (Imai et al. 2004).
As our patient has only thrombocytopenia, mild eczema with no infections or evidence of immune dysregulation he was assigned severity score of 2 according to the scoring system suggested by Zhu et al. (1997) Gene therapy has focused on patients with severity score from 3–5 who display a WAS phenotype characterized by bleeding, severe eczema, and severe infections (Abina et al. 2015).
Mutations in the WAS gene result in a broad range of disease severity and can be divided in different groups according to their effect on WASp expression. Mutations include null mutations (nonsense mutations, deletions, insertions with frameshift) - 46%, missense mutations - 42%, and splice anomalies - 12%. As previously published, most missense mutations are located in exons 1 through 4, in the WH1 domain resulting in an XLT phenotype or low severity WAS score, while nonsense mutations were observed as causing a severe phenotype (Imai et al. 2003, 2004). The genotype-phenotype correlation in WAS is not absolute. Other published data failed to demonstrate a clear correlation of missense mutations genotype/protein expression and phenotype, with only half of the patients carrying missense mutations exhibiting the XLT phenotype and detectable WASp (Liu et al. 2015). Clinicians often rely on published case report series to determine prognosis, given the wide clinical spectrum of WAS. Thus, it is important to broaden the genotypic and phenotypic spectrum of WAS mutations. We have shown a novel mutation causing a mild phenotype of WAS/XLT, our findings also support the notion of genetic testing in patients with thrombocytopenia and mild symptoms to ensure an early diagnosis of inborn errors of immunity.
REFERENCES
Abina S.H.-B., Gaspar H.B., Blondeau J., Caccavelli L., Charrier S., Buckland K., Picard C., Six E., Himoudi N., Gilmour K., McNicol A.M., Hara H., Xu-Bayford J., Rivat C., Touzot F., Mavilio F., Lim A., Treluyer J.M., Héritier S., Lefrère F., Magalon J., Pengue-Koyi I., Honnet G., Blanche S., Sherman E.A., Male F., Berry C., Malani N., Bushman F.D., Fischer A., Thrasher A.J., Galy A., and Cavazzana M. 2015. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. J. Am. Med. Assoc. 313(15): 1550–1563.
Albert M.H., Bittner T.C., Nonoyama S., Notarangelo L.D., Burns S., Imai K., Espanol T., Fasth A., Pellier I., Strauss G., Morio T., Gathmann B., Noordzij J.G., Fillat C., Hoenig M., Nathrath M., Meindl A., Pagel P., Wintergerst U., Fischer A., Thrasher A.J., Belohradsky B.H., and Ochs H.D. 2010. X-linked thrombocytopenia (XLT) due to WAS mutations: Clinical characteristics, long-term outcome, and treatment options. Blood, 115(16): 3231–3238.
Buchbinder D., Nugent D.J., and Illipovich A.H. 2014. Wiskott-Aldrich syndrome: Diagnosis, current management, and emerging treatments. Appl. Clin. Genet. 7: 55–66.
Chou H.-C., Antón I.M., Holt M.R., Curcio C., Lanzardo S., Worth A., Burns S., Thrasher A.J., Jones G.E., and Calle Y. 2006. Report WIP regulates the stability and localization of WASP to podosomes in migrating dendritic cells. Curr. Biol. 16: 2337–2344.
Dupuis-Girod S., Medioni J., Haddad E., Quartier P., Cavazzana-Calvo M., Deist F.L., Basile G.S., Delaunay J., Schwarz K., Casanova J.-L., Blanche S., and Fischer A. 2003. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics, 111(5): e622–e627.
Imai K., Nonoyama S., and Ochs H.D. 2003. WASP (Wiskott-Aldrich syndrome protein) gene mutations and phenotype. Curr. Opin. Allergy Clin. Immunol. 3(6): 427–436.
Imai K., Morio T., Zhu Y., Jin Y., Itoh S., Kajiwara M., Yata J., Mizutani S., Ochs H.D., and Nonoyama S. 2004. Clinical course of patients with WASP gene mutations. Blood, 103(2): 456–464.
Kim A.S., Kakalis L.T., Abdul-Manan N., Liu G.A., and Rosen M.K. 2000. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature, 404(6774): 151–158.
Kirchhausen T. and Rosen F.S. 1996. Disease mechanism: Unravelling Wiskott-Aldrich syndrome. Curr. Biol. 6(6): 676–678.
Liu D., Zhang Z., Zhao Q., Jiang L., Liu W., and Tu W. 2015. Wiskott – Aldrich Syndrome/X-Linked Thrombocytopenia in China : Clinical Characteristic and genotype – phenotype correlation. Pediatr. Blood Cancer. 62(9): 1601–1608.
Luthi J.N., Gandhi M.J., and Drachman J.G., 2003. X-linked thrombocytopenia caused by a mutation in the Wiskott-Aldrich syndrome (WAS) gene that disrupts interaction with the WAS protein (WASP)-interacting protein (WIP). Exp. Hematol. 31(2): 150–158.
Massaad M.J., Ramesh N., and Geha R.S. 2013. Wiskott-Aldrich syndrome: A comprehensive review. Ann. N. Y. Acad. Sci. 1285(1): 26–43.
Perry G.S., Spector B.D., Schuman L.M., Mandel J.S., Anderson V.E., McHugh R.B., Hanson M.R., Fahlstrom S.M., Krivit W., and Kersey J.H. 1980. The Wiskott-Aldrich syndrome in the United States and Canada (1892–1979). J. Pediatr. 97(1): 72–78.
Rivers E., Thrasher A.J., Rivers E., and Thrasher A.J. 2017. Wiskott-Aldrich syndrome protein : Emerging mechanisms in immunity. Eur. J. Immunol. 47: 1857–1866.
Scherl A., Couté Y., Déon C., Callé A., Kindbeiter K., Sanchez J-C., Greco A., Hochstrasser D., and Diaz J.-J. 2002. Functional proteomic analysis of human nucleolus. Mol. Biol. Cell, 13(11): 4100–4109.
Zhu Q., Watanabe C., Liu T., Hollenbaugh D., Blaese R.M., Kanner S.B., Aruffo A., and Ochs H.D. 1997. Wiskott-Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression, and phenotype. Blood, 90(7): 2680–2689.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 8 • Number 3 • September 2021
Pages: 94 - 98
History
Received: 9 August 2021
Accepted: 17 August 2021
Accepted manuscript online: 20 August 2021
Copyright
© 2021.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
JennyGarkaby and JuliaUpton. 2021. Wiskott-Aldrich syndrome caused by a novel mutation in the WAS gene and presenting with a mild phenotype. LymphoSign Journal.
8(3): 94-98. https://doi.org/10.14785/lymphosign-2021-0022
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Get Access
Login options
Check if you access through your login credentials or your institution to get full access on this article.
