Hemophagocytic lymphohistiocytosis associated with ataxia telangiectasia
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening inflammatory condition believed to be caused by uncontrolled activation of macrophages and histiocytes. HLH may be triggered by infections or associated with malignancy, metabolic disorders and drug toxicity, or alternatively, by a variety of genetic defects. While this disorder has been reported to be associated with a growing number of primary immunodeficiencies, especially those with significant T cell and (or) NK cell dysfunction, it has never been reported in ataxia telangiectasia (AT). AT is characterized by truncal ataxia, dilatation of blood vessels, immunodeficiency and a high predisposition to cancer. Almost all cases of AT have at least 1 or a combination of more than 1 of the following features: low immunoglobulin levels, inability to produce specific antibodies in response to vaccination, T cell lymphopenia and (or) T cell dysfunction. In this report, we describe the first case of a fatal episode of HLH in a patient with AT. The overlapping laboratory anomalies of HLH and lymphoid malignancy poses a challenge for accurate diagnosis, and awareness of the phenomenon by clinicians may result in earlier treatment and resolution of inflammation.
Statement of novelty: HLH can affect various types of immunodeficiency but has never been reported in patients with AT. Here, we report the first case of a fatal episode of HLH in a patient with AT.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening inflammatory condition believed to be caused by uncontrolled activation of macrophages and histocytes (Janka 2007). HLH may be triggered by infections or associated with malignancy, metabolic disorders and drug toxicity (Henter et al. 1991; Janka et al. 1998), or alternatively, by a variety of genetic defects.
The familial form of HLH presents frequently in infants born to consanguineous parents and may involve a history of other affected family members. Most patients in this group carry lesions in genes that are responsible for the cytotoxic function of T cells and NK cells. Commonly, genetic mutations in PRF-1, UNC13D, or STXII, encoding for cytolytic granule trafficking and exocytosis, are detected in these patients (Goransdotter Ericson et al. 2001; Feldmann et al. 2003; zur Stadt et al. 2005).
HLH was also reported to be associated with a growing number of primary immunodeficiencies, especially those with significant T cell and (or) NK cell dysfunction (Egeler et al. 1996; Dalal et al. 2015).
Ataxia telangiectasia (AT) is an autosomal recessive disorder characterized by truncal ataxia, dilatation of blood vessels, immunodeficiency and a high predisposition to cancer (Boder and Sedgwick 1958; Micol et al. 2011). Patients present early in life with hypotonia and delayed developmental milestones, and are frequently diagnosed with cerebral palsy. Later in childhood, ataxia becomes evident as well as telangiectasia in the eyes and ear lobes. Patients may suffer recurrent pulmonary infections and malignancy. Immune abnormalities include immunoglobulin deficiencies, T cell lymphopenia and dysfunction, and a dysplastic thymus (Nowak-Wegrzyn et al. 2004; Driessen et al. 2013).
Typical laboratory features consist of increased alpha-fetoprotein, increased chromosomal breakage and increased sensitivity to ionizing radiation (Canman and Lim 1998; Suzuki et al. 1999; Wu et al. 2000; Stray-Pedersen et al. 2007). AT is caused by mutations in the ATM gene, which is involved in DNA repair during double-stranded breaks (Savitsky et al. 1995; Staples et al. 2008).
HLH can affect various types of immunodeficiency but has never been reported in patients with AT. Here, we report the first case of a fatal episode of HLH in a patient with AT.
Case report
The patient was born at term to consanguineous parents of Italian descent. Development was delayed and a neurological assessment revealed hypotonia and poor head control. Sitting and standing were also markedly delayed and he was given a diagnosis of mild cerebral palsy. He was unstable when he walked and his gait was wide. At the age of 4 years, he was diagnosed with ataxia and was prescribed physiotherapy.
AT was suspected at the age of 6 years when he suffered from dysarthria, ataxia and swallowing difficulties, and was found to have dilated blood vessels in his eyes and ear lobes. Diagnosis was confirmed by the finding of markedly elevated alpha-fetoprotein as well as increased chromosomal breakage.
He continued to deteriorate neurologically and was wheelchair bound by the age of 9 years. At the age of 7 years, he suffered a bout of pneumonia and subsequently developed chronic bronchitis. At the age of 11 years, he developed persistent fevers with no signs of a source of infection. He gradually stopped eating and was admitted for evaluation for a possible malignancy. He was found to have an enlarged liver and spleen as well as lymphadenopathy. Laboratory evaluation revealed increased sedimentation rate, increased lactate dehydrogenase and ferritin, low platelets of 65 × 103/µL and reduced neutrophil count at 1.2 × 103/µL. This combination of clinical manifestations and laboratory abnormalities raised the possibility of a lymphoid malignancy, especially in a patient with AT.
A bone marrow biopsy was therefore performed. While no malignant cells were detected, a large number of hemophagocytic macrophages were identified (Figure 1). This finding supported the diagnosis of HLH. In fact, the clinical features of splenomegaly and laboratory aberrations, including 2 lineage cytopenia, hypertriglyceridemia, increased ferritin as well as typical histopathological changes, meet the criteria for the diagnosis of HLH (Table 1).
Figure 1:
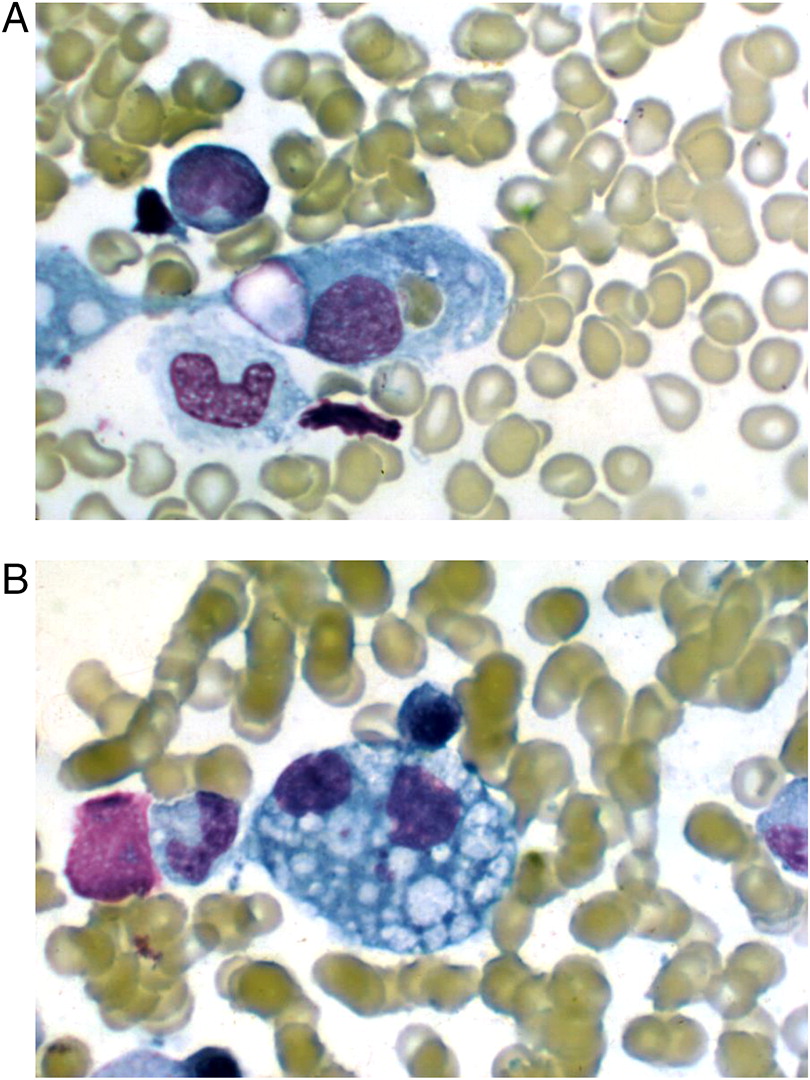
Table 1:
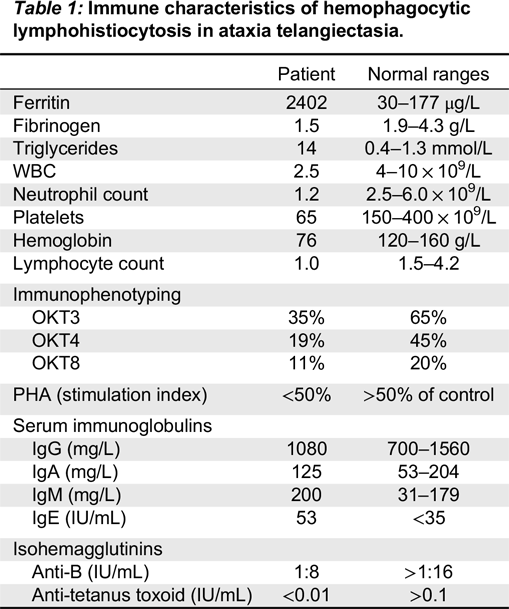
The patient was initially treated with broad spectrum antibiotics with little effect, and was subsequently switched to dexamethasone once HLH was suspected. Unfortunately, within 4 days he developed multi-organ failure leading to death.
Discussion
The symptoms of HLH consist of persistent fever, hepatosplenomegaly, multi-lineage cytopenia, morbiliforme rash, encephalopathy and seizures (Henter et al. 2007). Indeed, our patient presented with most of these typical features. Without treatment, familial HLH is fatal in more than 85% of cases within 1 year of diagnosis (Janka 1983). Even specific therapy cannot rescue up to 45% of cases. Treatment is aimed at suppressing inflammation by using immunosuppressive agents such as dexamethasone, etoposide and cyclosporine, and when appropriate, bone marrow transplantation (Henter et al. 2002). In spite of immunosuppression therapy, our patient deteriorated rapidly and died of multi-organ failure.
It is surprising that HLH was not reported previously in patients with AT. Almost all AT cases have at least 1 or a combination of more than 1 of the following features: low immunoglobulin levels, inability to produce specific antibodies in response to vaccination, T cell lymphopenia and (or) T cell dysfunction. Some patients present with profound T cell deficiency and are detected by newborn screening for SCID (Puck 2012). This may possibly be explained by the dysfunction of ATM in repairing rearrangement of the T cell receptor gene.
This case demonstrates that some overlapping laboratory anomalies of HLH and lymphoid malignancy may pose a challenge for an accurate diagnosis. It is therefore possible that, if suspected, HLH may be a more common manifestation in AT than expected. This knowledge may alert clinicians, resulting in earlier treatment and resolution of inflammation.
REFERENCES
Boder E. and Sedgwick R.P. 1958. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics. 21:526–554.
Canman C.E. and Lim D.S. 1998. The role of ATM in DNA damage responses and cancer. Oncogene. 17:3301–3308.
Dalal B.I., Vakil A.P., Khare N.S., Wang S.Y., Richards M.J., and Chen L.Y. 2015. Abnormalities of the lymphocyte subsets and their immunophenotype, and their prognostic significance in adult patients with hemophagocytic lymphohistiocytosis. Ann. Hematol. 94:1111–1117.
Driessen G.J., Ijspeert H., Weemaes C.M., Haraldsson A., Trip M., Warris A., van der Flier M., Wulffraat N., Verhagen M.M., Taylor M.A., van Zelm M.C., van Dongen J.J., van Deuren M., and van der Burg M. 2013. Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T-cell homeostasis and reduced immune repertoire diversity. J. Allergy Clin. Immunol. 131:1367–1375.e9.
Egeler R.M., Shapiro R., Loechelt B., and Filipovich A. 1996. Characteristic immune abnormalities in hemophagocytic lymphohistiocytosis. J. Pediatr. Hematol. Oncol. 18:340–345.
Feldmann J., Callebaut I., Raposo G., Certain S., Bacq D., Dumont C., Lambert N., Ouachée-Chardin M., Chedeville G., Tamary H., Minard-Colin V., Vilmer E., Blanche S., Le Deist F., Fischer A., and de Saint Basile G. 2003. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 115:461–473.
Goransdotter Ericson K., Fadeel B., Nilsson-Ardnor S., Soderhall C., Samuelsson A., Janka G., Schneider M., Gurgey A., Yalman N., Revesz T., Egeler R., Jahnukainen K., Storm-Mathiesen I., Haraldsson A., Poole J., de Saint Basile G., Nordenskjold M., and Henter J. 2001. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am. J. Hum. Genet. 68:590–597.
Henter J.I., Elinder G., Soder O., and Ost A. 1991. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr. Scand. 80:428–435.
Henter J.I., Horne A., Arico M., Egeler R.M., Filipovich A.H., Imashuku S., Ladisch S., McClain K., Webb D., Winiarski J., and Janka G. 2007. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer. 48:124–131.
Henter J.I., Samuelsson-Horne A., Arico M., Egeler R.M., Elinder G., Filipovich A.H., Gadner H., Imashuku S., Komp D., Ladisch S., Webb D., and Janka G. 2002. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 100:2367–2373.
Janka G., Imashuku S., Elinder G., Schneider M., and Henter J.I. 1998. Infection- and malignancy-associated hemophagocytic syndromes: Secondary hemophagocytic lymphohistiocytosis. Hematol. Oncol. Clin. North Am. 12:435–444.
Janka G.E. 1983. Familial hemophagocytic lymphohistiocytosis. Eur. J. Pediatr. 140:221–230.
Janka G.E. 2007. Familial and acquired hemophagocytic lymphohistiocytosis. Eur. J. Pediatr. 166:95–109.
Micol R., Ben Slama L., Suarez F., Le Mignot L., Beauté J., Mahlaoui N., Dubois d’Enghien C., Laugé A., Hall J., Couturier J., Vallée L., Delobel B., Rivier F., Nguyen K., Billette de Villemeur T., Stephan J.L., Bordigoni P., Bertrand Y., Aladjidi N., Pedespan J.M., Thomas C., Pellier I., Koenig M., Hermine O., Picard C., Moshous D., Neven B., Lanternier F., Blanche S., Tardieu M., Debré M., Fischer A., and Stoppa-Lyonnet D. 2011. Morbidity and mortality from ataxia-telangiectasia are associated with ATM genotype. J. Allergy Clin. Immunol. 128:382–389.e1.
Nowak-Wegrzyn A., Crawford T.O., Winkelstein J.A., Carson K.A., and Lederman H.M. 2004. Immunodeficiency and infections in ataxia-telangiectasia. J. Pediatr. 144:505–511.
Puck J.M. 2012. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: The winner is T-cell receptor excision circles. J. Allergy Clin. Immunol. 129:607–616.
Savitsky K., Sfez S., Tagle D.A., Ziv Y., Sartiel A., Collins F.S., Shiloh Y., and Rotman G. 1995. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Mol. Genet. 4:2025–2032.
Staples E.R., McDermott E.M., Reiman A., Byrd P.J., Ritchie S., Taylor A.M., and Davies E.G. 2008. Immunodeficiency in ataxia telangiectasia is correlated strongly with the presence of two null mutations in the ataxia telangiectasia mutated gene. Clin. Exp. Immunol. 153:214–220.
Stray-Pedersen A., Borresen-Dale A.L., Paus E., Lindman C.R., Burgers T., and Abrahamsen T.G. 2007. Alpha fetoprotein is increasing with age in ataxia–telangiectasia. Eur. J. Paediatr. Neurol. 11:375–380.
Suzuki K., Kodama S., and Watanabe M. 1999. Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J. Biol. Chem. 274:25571–25575.
Wu X., Ranganathan V., Weisman D.S., Heine W.F., Ciccone D.N., O’Neill T.B., Crick K.E., Pierce K.A., Lane W.S., Rathbun G., Livingston D.M., and Weaver D.T. 2000. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 405:477–482.
zur Stadt U., Schmidt S., Kasper B., Beutel K., Diler A.S., Henter J.I., Kabisch H., Schneppenheim R., Nurnberg P., Janka G., and Hennies H.C. 2005. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum. Mol. Genet. 14:827–834.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 4 • Number 3 • September 2017
Pages: 113 - 116
History
Received: 7 July 2017
Accepted: 27 August 2017
Accepted manuscript online: 29 August 2017
Copyright
© 2017.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
MohammadAlsalamah and Chaim M.Roifman. 2017. Hemophagocytic lymphohistiocytosis associated with ataxia telangiectasia. LymphoSign Journal.
4(3): 113-116. https://doi.org/10.14785/lymphosign-2017-0007
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Get Access
Login options
Check if you access through your login credentials or your institution to get full access on this article.
