CD40 deficiency: a unique adult patient with hyper immunoglobulin M syndrome and normal expression of CD40
Abstract
Background: CD40 deficiency is an autosomal recessive, combined primary immunodeficiency characterized by defects of immunoglobulin class switch recombination and somatic hypermutation. It is part of an expanding group of diseases collectively known as hyper immunoglobulin M syndromes. Clinical manifestations of the disease usually begin early in life with recurrent sinopulmonary bacterial infections and susceptibility to opportunistic organisms. Only 16 patients from 12 unrelated families have been reported to date, all with lack of membrane expression of CD40 molecule.
Methods: Prospective and retrospective data was collected from the patient’s medical records, and Sanger sequencing, flow cytometry analysis, real-time polymerase chain reaction and western blotting were performed.
Results: In contrast with the patients reported previously, our patient’s mutation permits CD40 expression on the cell membrane and adds 37 amino acids to the cytoplasmic domain of the protein. We predict this change to affect 1 of the 2 known TRAF2 binding sites, as well as generate defective internalization of the receptor, both of which are required processes for functional signaling by CD40.
Conclusion: Our patient’s unique phenotype is an opportunity to further understand the biology and function of the CD40 receptor. As illustrated by this case, relying solely on flow cytometry for diagnosis of CD40 deficiency has the potential of overlooking patients with mutations that may allow residual protein expression. Therefore, confirmatory mutation analysis should always be performed.
Statement of novelty: We report on a patient with a novel mutation in CD40 and a unique phenotype, characterized by a complete lack of CD40 function despite normal protein expression. To our knowledge, this has not been reported previously. The patient has a milder phenotype than described for other patients with CD40 deficiency.
Introduction
CD40 deficiency is a rare autosomal recessive combined primary immunodeficiency characterized by defects of immunoglobulin (Ig) class switch recombination (CSR) and somatic hypermutation (SHM), and is part of an expanding group of genetic disorders collectively known as hyper immunoglobulin M (HIGM) syndromes. Some of these diseases result from intrinsic defects in B cells and generate a pure humoral immunodeficiency; others, like CD40 and CD40 ligand deficiency, result in a clinical picture of combined immunodeficiency as they also affect the function of cells such as monocytes, macrophages and dendritic cells, consequently affecting T cell-mediated responses (Davies and Thrasher 2010).
CD40 deficiency has only been reported in 16 patients from 12 unrelated families since its description in 2001 (Ferrari et al. 2001; Kutukculer 2003a, 2003b; Mazzolari et al. 2007; Al-Dhekri et al. 2012; Al-Saud et al. 2013; Mishra et al. 2015). Here, we present an additional and unique patient with the disease. CD40, a member of the tumor necrosis factor receptor (TNFR) family, is expressed constitutively on the surface of B cells, monocytes, macrophages, dendritic cells, T cells, and other non-immune cell types such as epithelial, epidermal, endothelial, thymic epithelial, fibroblast, glial, and neuronal cells. CD40 binds to CD40 ligand (CD40L, CD154, or TNFSF5), which is predominately and transiently expressed on activated CD4+ T cells (Vogel and Noelle 1998; Lougaris et al. 2005; Notarangelo et al. 2007; Davies and Thrasher 2010). CD40/CD40 ligand interaction is an absolute requirement to initiate CSR and SHM by promoting germinal center development in secondary lymphoid organs (Notarangelo et al. 2006).
Following cross linking of CD40 on the B cell membrane, intracellular signaling is initiated through the activation of TNF receptor associated factors (TRAFs), particularly TRAF-2, -3, -5, and -6. These factors function as adaptor molecules to recruit different kinases (e.g., mitogen-activated protein kinases or MAPKs, extracellular signal-regulated kinase or ERK, phospholipase Cγ2, syk, and phosphatidylinositol 3) resulting in activation of the transcription factors: activator protein-1 (AP-1) and NFκB (Lougaris et al. 2005; Notarangelo et al. 2006; Lanzi et al. 2010). Signaling mediated by CD40 might also involve pathways independent of TRAFs, such as activation of Janus-associated kinases (JAKs), particularly JAK3, and also signal transducers and activators of transcription (STATs), mainly STAT6 and STAT3 (Lougaris et al. 2005).
CD40 deficient patients display an impaired production of IgG, IgA, IgE, and normal or elevated levels of IgM. They have no antibody response to protein antigens but can have some IgM anti-polysaccharide antibodies. These patients also have absent or very low numbers of circulating memory IgD−CD27+ B cells and abnormal lymph nodes with primary follicles but absent germinal centers (Notarangelo et al. 2007). Patients usually present in the first months of life with susceptibility to recurrent sinopulmonary bacterial infections. However, and given the expression of CD40 on activated monocytes and dendritic cells and the linkage with CD40 ligand on T helper cells, these patients are also susceptible to infections by opportunistic intracellular pathogens such as Pneumocystis jiroveci causing pneumonia and Cryptosporidium species causing chronic gastrointestinal infections (Lougaris et al. 2005). Seven out of the 16 patients with CD40 deficiency reported to date have shown significant neutropenia (Lougaris et al. 2005; Mazzolari et al. 2007; Al-Saud et al. 2013), and so far none of the CD40 deficient patients have developed signs of malignancy.
Diagnosis is commonly made by demonstrating the absent expression of CD40 at the surface of B cells and monocytes by flow cytometry. All of the patients described to date have a complete lack of expression of the CD40 protein.
Treatment is similar to that of combined immunodeficiency with the prevention of both bacterial and opportunistic infections. Intravenous immunoglobulin (IVIG) replacement and prophylaxis against P. jiroveci with cotrimoxazole should be initiated at the time of diagnosis. Prevention of infection by Cryptosporidium relies mostly on hygienic measures. Hematopoietic stem cell transplantation (HSCT) for CD40 deficiency is not a complete cure as it will only correct the immunological manifestations by restoring the expression of CD40 on hematopoietic stem cell-derived cell lineages. Other cells, such as endothelial cells or neurons, will continue to be defective of this protein. Nevertheless, HSCT has been attempted in 4 patients, with successful immune reconstitution in 3 (Kutukculer 2003a, 2003b; Mazzolari et al. 2007; Al-Dhekri et al. 2012). Given the ubiquitous presence of CD40 among different cell types, gene therapy is not a foreseeable option, at least at this point in time.
Case report
We describe a unique adult patient with a novel mutation in CD40 allowing normal constitutive and induced expression of the protein in B cells and monocytes. The patient presented with susceptibility to infections at a later age and had a milder phenotype as compared to previously reported patients.
The patient is currently 21 years old. He was born to Iranian and French-Canadian non-consanguineous parents. He first presented to our clinic at the age of 15 years with a history of 3 documented pneumonias and 1 episode of septic arthritis, all presenting after he was 10 years of age. He had a history of isolated episodes of acute otitis media between the ages of 1 and 4 years that had resolved with oral antibiotics. He was healthy until the age of 10 years when he had the first episode of pneumonia. The second and third pneumonias occurred at the ages of 12 and 13 years, respectively. Also at the age of 13 years, he experienced an episode of right knee septic arthritis. He recovered fully from all of these infections after completing adequate antibiotic courses. He was also followed by an endocrinologist for short stature due to growth hormone deficiency, and received replacement therapy until the age of 15 years. When he was assessed in our service he was found to have undetectable (<0.1 g/L) titers for both IgG and IgA (normal ranges of 4.5–14.3 and 0.2–1.0 g/L, respectively) and elevated IgM of 19.2 g/L (0.2–1.8 g/L). He lacked protective titers of specific antibodies for vaccines despite having a complete immunization schedule. He had normal numbers of B cells and T cells (CD19+ 454, CD3+ 2526, CD4+ 1670, CD8+ 721, NK cells 161) and his lymphocyte proliferation responses to both mitogens and antigens were also normal. He was commenced on monthly IVIG replacement therapy that has continued to the present day, without any further episodes of severe or recurrent infections. As there was never any evidence of opportunistic infections he was not started on prophylactic antibiotics. He has remained clinically well.
Methods
Patient data was compiled prospectively and retrospectively from medical records and was entered into the Canadian Centre for Primary Immunodeficiency Registry and tissue bank, which was approved by the SickKids Research Ethics board (protocol no. 1000005598). This includes informed consent from the patient for genetic analysis and the collection of blood samples for this study. The patient was diagnosed and followed clinically before adulthood at The Hospital for Sick Children, Toronto, ON, Canada.
Molecular analysis
Sanger sequencing was used to analyze CD40 ligand, activation-induced cytidine deaminase (AID), and CD40 gene sequences. Patient’s genomic DNA was extracted from peripheral blood lymphocytes using the Geneaid Genomic DNA Mini Kit. Genomic DNA was amplified by PCR with specific primers designed upstream and downstream of the RNU4ATAC gene. Sequencing was done using GenomeLab Dye Terminator Cycle Sequencing (DTCS) Quick Start Kit (Beckman Coulter) and analyzed on CEQ 8000 Genetic Analysis System (Beckman Coulter).
Flow cytometry analysis
Lymphocyte immunophenotyping and expression of CD40, CD40 ligand, CD95, CD54, and CD86 were measured by flow cytometry on a single laser, 3 colour Coulter EPICS V Flow Cytometer from Beckman Coulter, CA (USA). All fluorescent labeled antibodies were purchased from eBiosciences (San Diego, CA, USA).
T- and B-cell proliferative responses
Lymphocyte proliferative responses to mitogens and to a panel of recall antigens were determined by thymidine incorporation at day 3 or day 6. All assays were performed in triplicate and were compared with simultaneously stimulated random normal controls.
Immunoglobulin and specific antibody determinations
Serum concentrations of immunoglobulins were measured by nephelometry. Serum IgE was measured by radioimmunoassay with the IgE PRIST kit (Pharmacia Diagnostics, Quebec, QC, Canada). Levels of specific antibodies were measured by ELISA.
Real-time polymerase chain reaction (RT-PCR)
RT-PCR was performed on an ABI 7500 real-time PCR system (Applied Biosystems).
Western blotting
Whole-cell lysates were prepared in a RIPA buffer and analyzed by Western blotting. Anti-RelA was purchased from Santa Cruz Biotechnology Inc. (TX, USA).
Results
Molecular analysis showed normal CD40 ligand and AID gene sequences. However, analysis of the CD40 gene revealed a homozygous 1 base pair deletion in exon 9 of both genomic and complementary DNA. The mutation c.823delG results in p.E275RfsX38 that translates into a frame shift adding 37 extra amino acids to the cytoplasmic domain of the protein (Figure 1).
Figure 1:
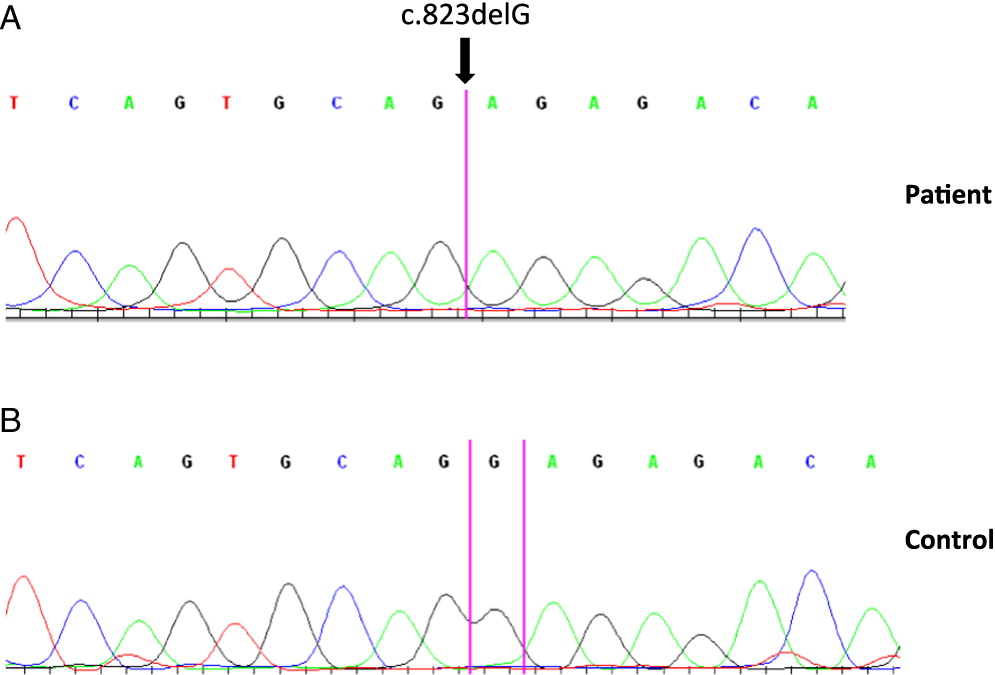
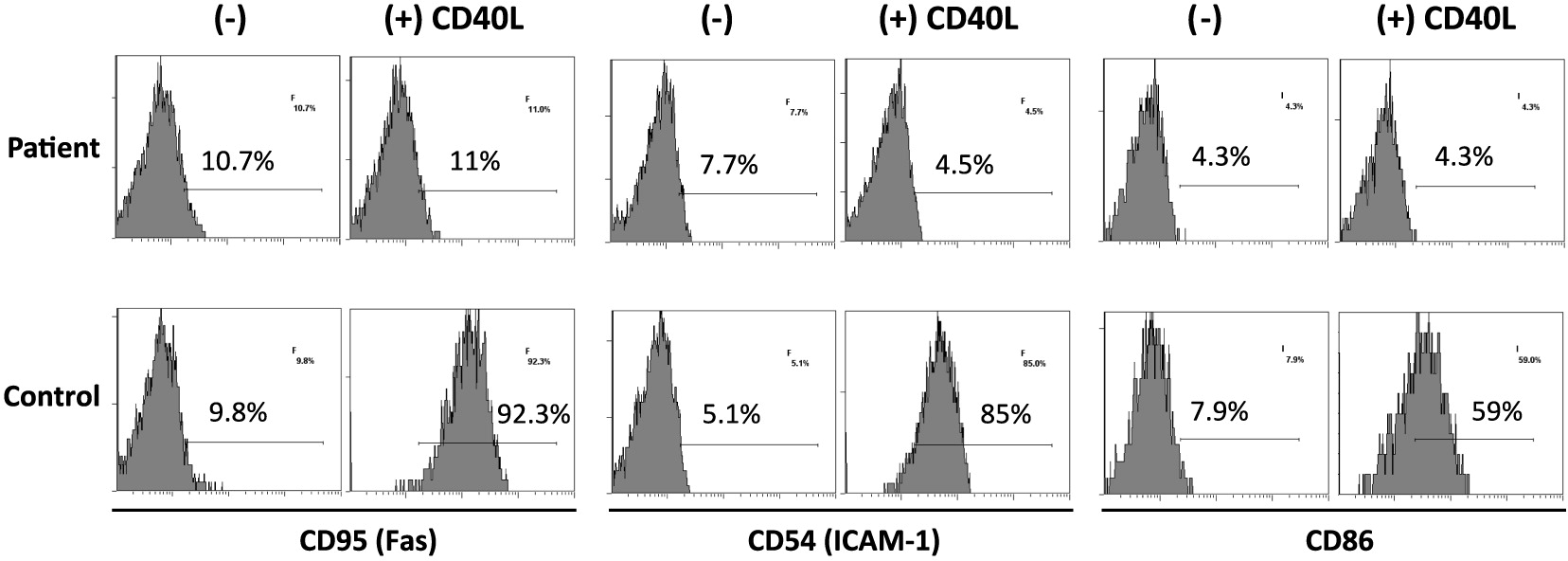
On further analysis by flow cytometry, we found that both his B cells and monocytes expressed CD40 normally on their surface, both constitutively and following induction with IFNγ (Figure 2). Despite normal CD40 surface expression, our patient showed defective immunoglobulin class switching with most of his B cells being naïve (IgM+IgD−) and an almost complete absence of the memory phenotype (IgD−CD27+) (Figure 3). Also, when cultured with recombinant CD40 ligand, our patient’s B cells failed to induce expression of CD95 (also known as Fas), integrin receptor CD54 (ICAM-1), and of co-stimulatory molecule CD86; all of which are important molecules in cell signaling (Figure 4). This conclusively demonstrates the lack of CD40 receptor function despite its normal expression on the cell surface, and has not been previously reported.
Figure 2:
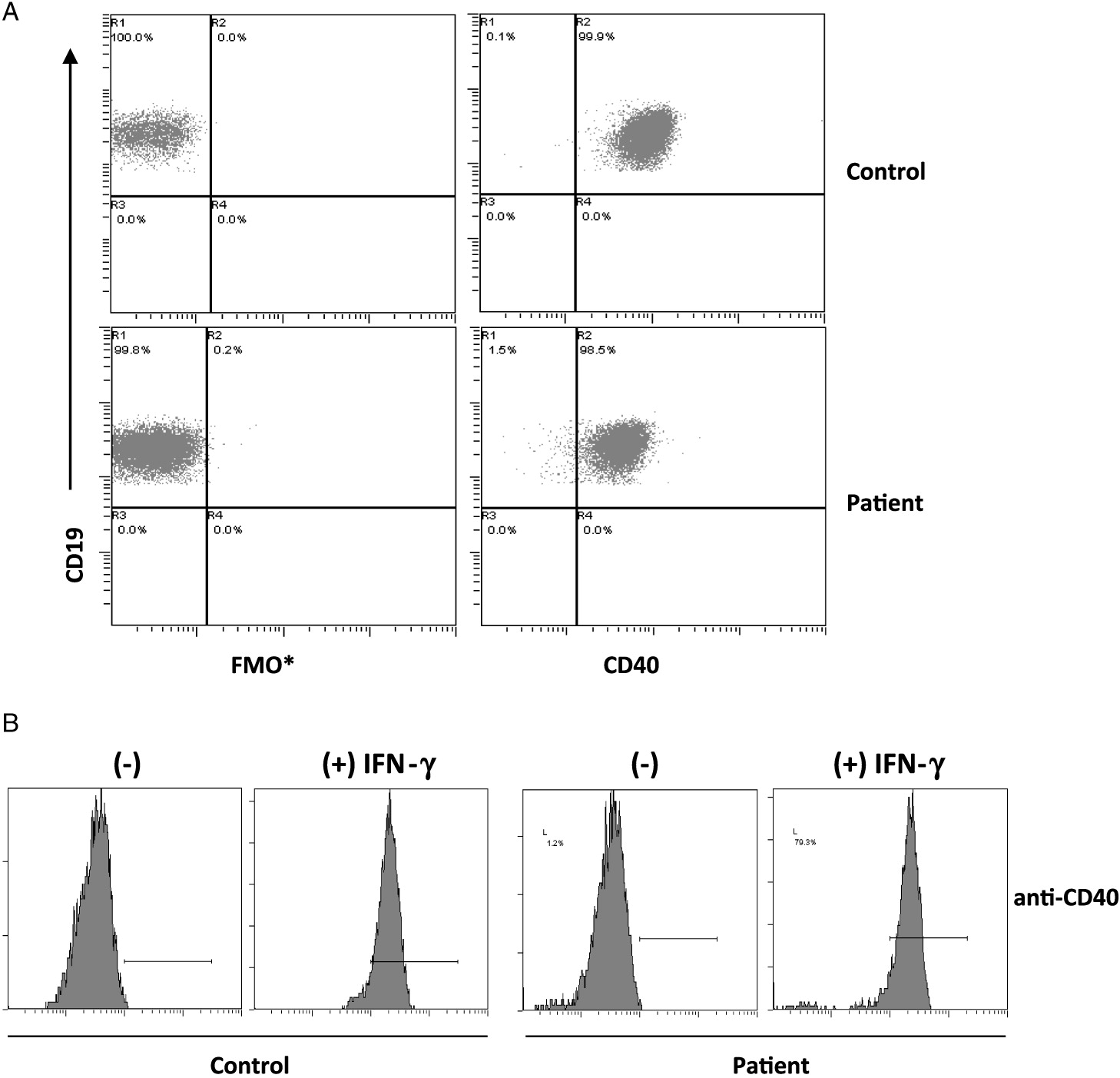
Figure 3:
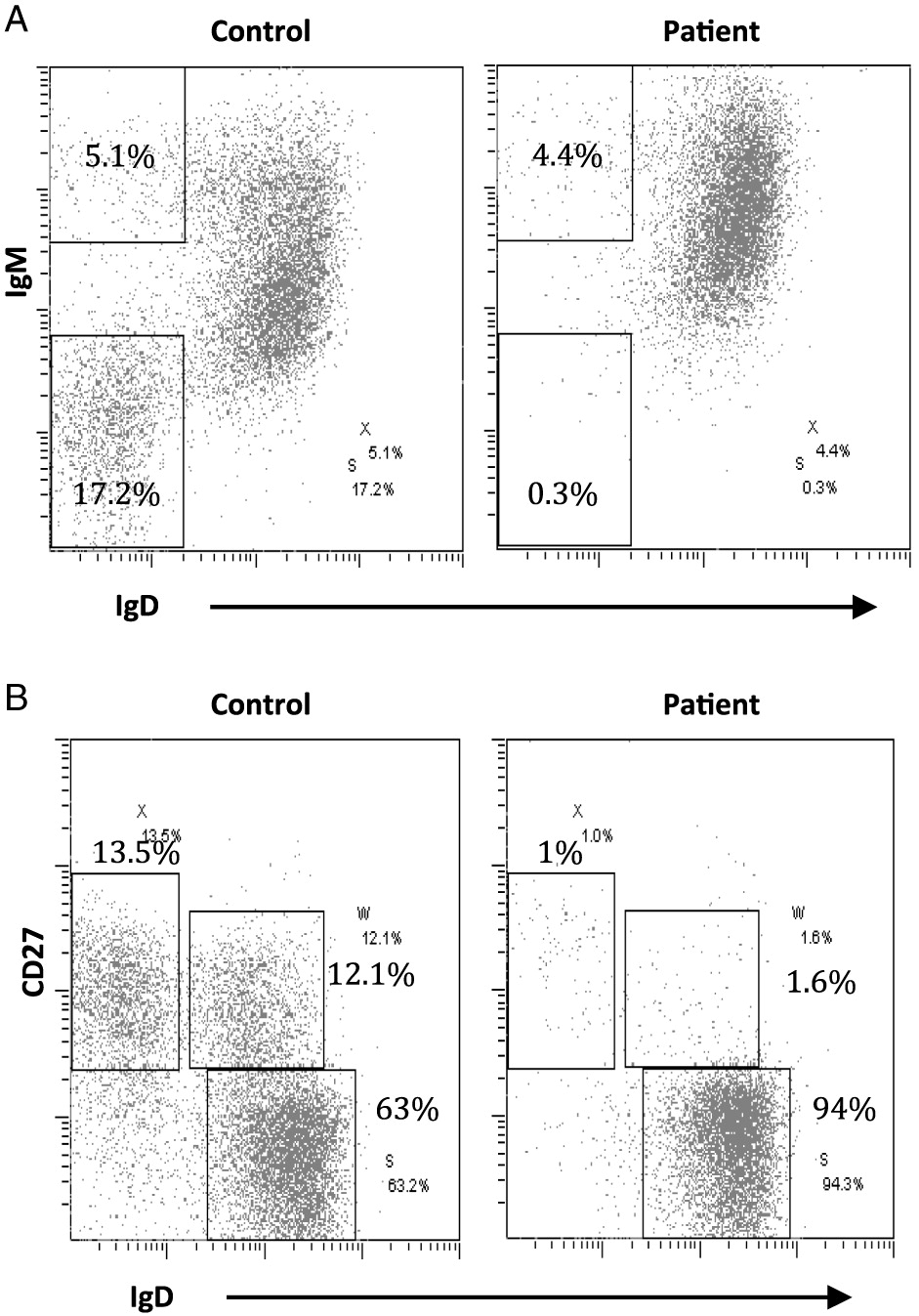
Figure 4:
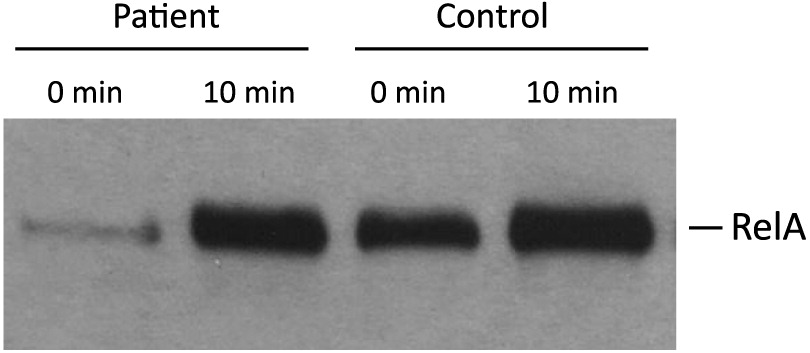
Another important observation is that the NFκB signaling pathway is functional in our patient, as shown by normal RelA nuclear translocation after stimulation with CD40 ligand. RelA is an important molecule for nuclear translocation and activation of NFκB (Figure 5).
Figure 5:
Moreover, we found lack of down-regulation of expression of CD40 after stimulation with CD40 ligand (data not shown), which likely suggests a problem with receptor internalization.
Discussion
As illustrated by our patient, relying only on flow cytometry for diagnosis of CD40 deficiency has the potential of overlooking patients with mutations that may allow some degree of protein expression, and therefore confirmatory mutation analysis should always be performed. To our knowledge, our patient represents the first reported case of CD40 deficiency with normal membrane expression of CD40. Missense mutations or small in-frame deletions in 4 of the reported patients with CD40 deficiency were able to generate mutated proteins. However, these were retained in the endoplasmic reticulum and thus not expressed in the cell membrane (Lanzi et al. 2010).
Based on the behavior of other similar receptors, we propose that the deletion of the terminal 3 amino acids and addition of an extra 37 amino acids to the intracellular tail interferes with normal CD40 receptor internalization, which is an absolute requirement for this receptor’s downstream signaling. Interestingly, and as we were also able to show, it is known from other receptors that NFκB activation can proceed without internalization. Alternatively, or perhaps in addition, we believe that the predicted loss of the terminal TRAF2 binding site may be critical for normal function of the receptor through the MAP kinase signaling pathway. Both normal internalization of the CD40 receptor and binding of MAP kinases to TRAF2 are required for functional signaling by CD40. More studies are required to fully elucidate these processes.
It is unclear why our patient presented with a milder phenotype, despite lack of function of CD40 even when it was expressed in the cell membrane. Perhaps the interaction of this extracellular domain with other immune and non-immune cells allows for some compensatory or alternative signaling, something that is yet to be understood.
Our patient has given us a unique opportunity to further understand the biology and function of the CD40 receptor. As more patients are described and, consequently, as the phenotype of the disease broadens and is better understood, we will learn more about this primary immunodeficiency that clearly goes beyond a defect of CSR.
Acknowledgements
We would like to thank our patient for allowing us to explore his immunodeficiency in more detail. This work was supported by the Canadian Centre for Primary Immunodeficiency, the Jeffrey Modell Foundation, and Immunodeficiency Canada.
REFERENCES
Al-Dhekri H., Al-Sum Z., Al-Saud B., Al-Mousa H., Ayas M., Al-Muhsen S., Arnaout R., Al-Seraihy A., Hawwari A., and Al-Ghonaium A.2012. Successful outcome in two patients with CD40 deficiency treated with allogeneic HCST. Clin. Immunol.143(1):96–98.
Al-Saud B.K., Al-Sum Z., Alassiri H., Al-Ghonaium A., Al-Muhsen S., Al-Dhekri H., Arnaout R., Alsmadi O., Borrero E., Abu-Staiteh A., Rawas F., Al-Mousa H., and Hawwari A.2013. Clinical, immunological, and molecular characterization of hyper-IgM syndrome due to CD40 deficiency in eleven patients. J. Clin. Immunol.33(8):1325–1335.
Davies E.G. and Thrasher A.J.2010. Update on the hyper immunoglobulin M syndromes. Br. J. Haematol.149(2):167–180.
Ferrari S., Giliani S., Isalaco A., Al-Ghonaium A., Soresina A.R., Loubser M., Avanzini M.A., Marconi M., Badolato R., Ugazio A.G., Levy Y., Catalan N., Durandy A., Tbakhi A., Notarangelo L.D., and Piebani A.2001. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc. Natl. Acad. Sci. USA.98(22):12614–12619.
Kutukculer N., Aksoylar S., Kansoy S., Cetingul N., and Notarangelo L.D.2003a. Outcome of hematopoietic stem cell transplantation in hyper-IgM syndrome caused by CD40 deficiency. J. Pediatr.143(1):141–142.
Kutukculer N., Moratto D., Aydinok Y., Lougaris V., Aksoylar S., Piebani A., Genel F., and Notarangelo L.D.2003b. Disseminated cryptosporidium infection in an infant with hyper-IgM syndrome caused by CD40 deficiency. J. Pediatr.142(2):194–196.
Lanzi G., Ferrari S., Vihinen M., Caraffi S., Kutukculer N., Schiaffonati L., Piebani A., Notarangelo L.D., Fra A.M., and Giliani S.2010. Different molecular behavior of CD40 mutants causing hyper-IgM syndrome. Blood. 116(26):5867–5874.
Lougaris V., Badolato R., Ferrari S., and Piebani A.2005. Hyper immunoglobulin M syndrome due to CD40 deficiency: Clinical, molecular, and immunological features. Immunol. Rev.203:48–66.
Mazzolari E., Lanzi G., Forino C., Lanfranchi A., Aksu G., Ozturk C., Gilliani S., Notarangelo L.D., and Kutukculer N.2007. First report of successful stem cell transplantation in a child with CD40 deficiency. Bone Marrow Transplant. 40(3):279–281.
Mishra A., Italia K., Gupta M., Desai M., and Madkaikar M.2015. Hyperimmunoglobulin syndrome due to CD40 deficiency: Possibly the first case from India. J. Postgrad. Med.61:46–48.
Notarangelo L.D., Lanzi G., Peron S., and Durandy A.2006. Defects of class-switch recombination. J. Allergy Clin. Immunol.117(4):855–864.
Notarangelo L.D., Lanzi G., Toniati P., and Giliani S.2007. Immunodeficiencies due to defects of class-switch recombination.Immunol. Res.38(1–3):68–77.
Vogel L.A. and Noelle R.J.1998. CD40 and its crucial role as a member of the TNFR family. Semin. Immunol.10(6):435–442.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 4 • Number 2 • June 2017
Pages: 70 - 76
History
Received: 9 April 2017
Accepted: 18 May 2017
Accepted manuscript online: 8 June 2017
Copyright
© 2017.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
LuisMurguia-Favela, NigelSharfe, ArianaKaranxha, AndreaBates, HarjitDadi, LorandCimpean, and Chaim M.Roifman. 2017. CD40 deficiency: a unique adult patient with hyper immunoglobulin M syndrome and normal expression of CD40. LymphoSign Journal.
4(2): 70-76. https://doi.org/10.14785/lymphosign-2017-0004
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Get Access
Login options
Check if you access through your login credentials or your institution to get full access on this article.
